
3种马铃薯病原菌多重PCR检测方法的建立
2021-01-09 08:34詹芳芳汤一凡
激光生物学报 2020年6期
詹芳芳,王 甜,汤一凡
(福建农林大学植物病毒研究所,福州 350002)
马铃薯(Solanum tuberosumL.)是世界范围内的重要粮食作物。尤其对发展中国家来说,马铃薯在充当营养丰富的食物、增加农户收入和保障粮食安全等方面发挥了不可替代的作用[1-2]。我国是马铃薯生产大国。2018年,我国马铃薯的种植面积和总产量分别为481.35万公顷和9 032.14万吨,均居世界首位[3]。然而,我国近十年来的马铃薯平均单产量仅为16.4吨/公顷,显著低于美国、欧洲和世界的平均单产量[4]。限制我国马铃薯单产水平提高的主要因素之一就是病害问题。
马铃薯容易受到多种病害的威胁,比如晚疫病(late blight)、早疫病(early blight)、黑痣病(black scurf)、枯萎病(fusarium wilt)、青枯病(bacterial wilt)和由马铃薯Y病毒(Potato virus Y, PVY)引起的病毒病等[5]。马铃薯晚疫病(potato late blight)的病原菌是致病疫霉(Phytophthora infestans),其能造成马铃薯茎叶死亡以及块茎腐烂,从而导致马铃薯毁灭性减产。目前,晚疫病仍然是对马铃薯生产威胁最大的病害,全球每年因晚疫病造成的经济损失高达60亿美元[6]。早疫病是仅次于晚疫病的第二大病害,主要由茄链格孢菌(Alternaria solani)引起,病情较轻时可导致马铃薯减产5%~10%,严重时可减产50%以上[7-8]。黑痣病也是一种对马铃薯生产造成严重威胁的世界性病害[9]。该病害的病原菌立枯丝核菌(Rhizoctonia solani)是一个复合种。根据菌丝融合、培养性状、致病性和DNA碱基同源性等特性,立枯丝核菌可划分为14个融合群(anastomosis groups, AGs),分别命名为AG-1~AG-13和AG-BI,侵染马铃薯的主要为AG-3融合群[10-11]。马铃薯黑痣病是一种土传病害,其病原菌一旦在土壤中定殖就很难根除,严重影响马铃薯的品质和产量,重症田块发病率可高达80%[12]。目前,针对这些病害的防治策略还是以选用抗病品种和化学防治为主[13-14],但它们在实践中往往不能有效地控制马铃薯病害的发生和流行[15-17]。因此,必须从源头上解决病害问题。在播种前对土壤或者种薯进行病原菌检测,能够有效控制病原菌的传播,从而减少和预防病害的流行。所以,开发快速、特异和灵敏的植物病原菌检测方法就显得尤为重要。
目前,基于聚合酶链式反应(polymerase chain reaction, PCR)的技术仍然是最常用的植物病原菌分子检测方法。PCR方法建立以后,随之衍生出了多种以PCR为基础的检测技术,包括多重PCR、实时荧光定量PCR(real-time PCR)、反转录PCR(reverse transcription PCR, RT-PCR)、巢式PCR、固相PCR等。多重PCR是在一个反应体系中同时扩增2个以上目的基因片段的PCR技术,具有节省时间、降低成本和提高效率的优势,其已经成为生命科学各个领域中的重要研究手段[18]。本研究针对马铃薯上的3种常见病原菌[即致病疫霉、茄链格孢菌和立枯丝核菌(AG-3融合群)]建立了一个多重PCR检测方法,通过1次多重PCR扩增就能同时检测出这3种病原菌。该检测方法能够节约时间,降低成本,提高检测效率,为马铃薯病害的早期预防、诊断及防控提供可靠的技术支持。
1 材料与方法
1.1 供试菌株
致病疫霉、立枯丝核菌(AG-3融合群)、辣椒疫霉(Phytophthora capsici)、链格孢菌(Alternaria alternata)、细极链格孢菌(Alternaria tenuissima)为本实验室保存和管理的菌株。大豆疫霉(Phytophthora sojae)由南京农业大学植物保护学院提供。茄链格孢菌购于中国农业微生物菌种保藏管理中心(Agricultural Culture Collection of China, ACCC),立枯丝核菌(AG-1融合群)和立枯丝核菌(AG-4融合群)由云南农业大学植物保护学院提供。
1.2 DNA提取
分别取适量供试病原菌的纯培养菌丝,冷冻抽干后打成菌丝粉,采用试剂盒(HP Fungal DNA kit, OMEGA)分别提取DNA。所得的DNA用BioDrop超微量蛋白核酸分析仪检测浓度和纯度后,置于-40℃保存备用。
1.3 引物设计
根据致病疫霉的elicitin基因INF1(GenBank登录号:AY830094.1)、立枯丝核菌(AG-3融合群)内毒素蛋白基因ENDO1(GenBank登录号:KM522918.1)和茄链格孢菌组蛋白基因(GenBank登录号:KF308960.1)的序列,应用引物设计和分析软件Premier 6.0分别设计3对特异性引物(表1),并委托上海生工生物工程有限公司合成。

表1 本研究中所使用的引物Tab. 1 Primers used in this study
1.4 单一PCR
PCR 反应体系(25.0 μL):Taq酶(TaKaRa, 5 U/μL)0.1 μL、10×PCR Buffer(100 mmol/L Tris-HCl,500 mmol/L KCl,15 mmol/L MgCl2)2.5 μL、dNTPs(Ta-KaRa,2.5 mmol/L)2.0 μL、DNA 模板1.0 μL、上游引物(10 μmol/L)1.0 μL、下游引物(10 μmol/L)1.0 μL,用ddH2O补足至25.0 μL。
扩增程序 :94℃预变性 3 min ;94℃变性 30 s,53℃退火30 s,72℃延伸30 s,35个循环 ;最后72℃延伸5 min。取5.0 μL产物进行1%的琼脂糖凝胶电泳,再用凝胶成像系统对产物条带进行分析并拍照记录。
1.5 多重PCR
1.5.1 引物浓度优化
多重PCR反应体系(25.0 μL):Taq酶(TaKaRa,5 U/μL)0.2 μL、10×PCR Buffer(100 mmol/L Tris-HCl,500 mmol/L KCl,15 mmol/L MgCl2)2.5 μL、dNTPs(Ta-KaRa,2.5 mmol/L)2.5 μL、3种DNA 模板各1.0 μL、上游引物(10 μmol/L)各0.4~0.6 μL、下游引物(10 μmol/L)各0.4~0.6 μL,用ddH2O补足至25.0 μL。
扩增程序 :94℃预变性 3 min ;94℃变性 30 s,53℃退火30 s,72℃延伸30 s,35个循环 ;最后72℃延伸5 min。取5.0 μL产物进行1%的琼脂糖凝胶电泳,再用凝胶成像系统对产物条带进行分析并拍照记录。
1.5.2 特异性验证
以致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌的基因组DNA为模板作为阳性对照,ddH2O为模板作为阴性对照,在最佳反应条件下,3对引物分别对致病疫霉、茄链格孢菌、立枯丝核菌(AG-3融合群)、大豆疫霉、辣椒疫霉、链格孢菌、细极链格孢菌、立枯丝核菌(AG-1融合群)和立枯丝核菌(AG-4融合群)的基因组 DNA 进行 PCR 扩增,以验证多重PCR检测方法的特异性。
1.5.3 灵敏度测定
分别按10倍梯度稀释3种DNA模板,得到3组质量浓度为1.0 ng/μL、100.0 pg/μL、10.0 pg/μL、1.0 pg/μL和0.1 pg/μL的DNA模板。在最佳的反应条件下进行多重PCR,以确定多重PCR检测方法的灵敏度。
1.5.4 土壤样品检测
在种植马铃薯的田块里采集土壤灭菌干燥,分别将致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌的菌丝按随机比例混合后加入土壤中,得到人工侵染阳性土壤样品。灭菌后的土壤为阴性土壤样品。采用土壤DNA提取试剂盒(DNeasy PowerSoil Pro Kit,Qiagen)提取土壤样品的DNA,再进行多重PCR检测以及琼脂糖凝胶电泳。
2 结果与分析
2.1 单一PCR结果
用设计好的3对引物(表1)分别对致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌的模板DNA进行单一PCR扩增,其琼脂糖凝胶电泳的结果显示(图1),茄链格孢菌的产物条带在100 ~250 bp之间,立枯丝核菌(AG-3融合群)的产物条带在250 bp的上方,致病疫霉的产物条带在500 bp的下方。该结果与设计的预期片段大小(209、307和401 bp)一致。
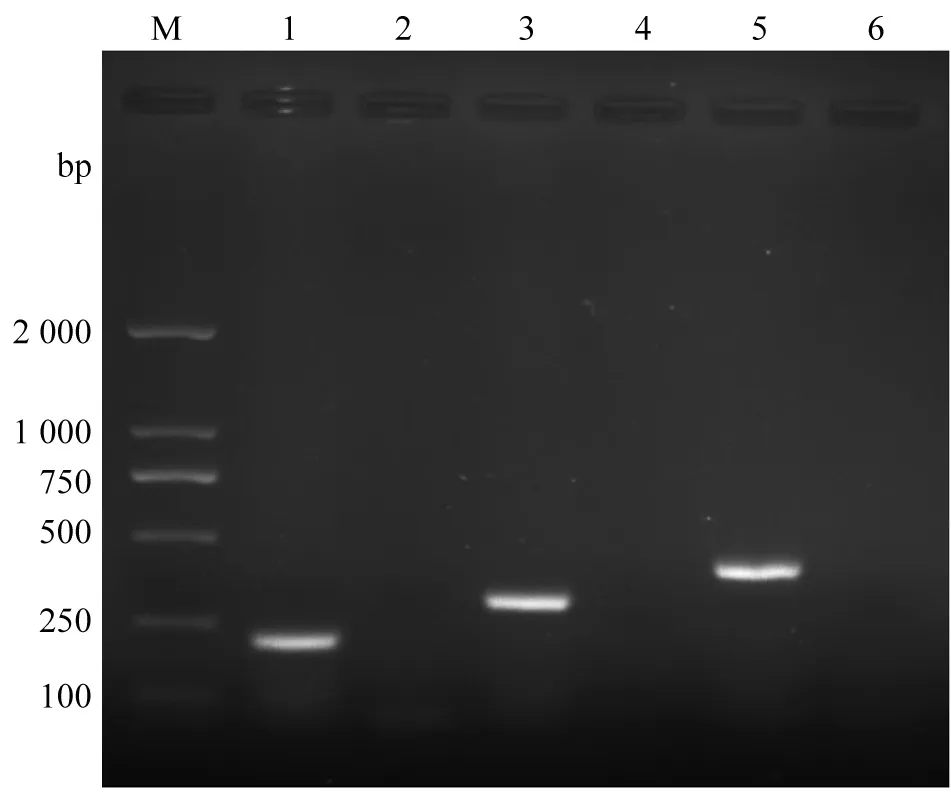
图1 单一PCR扩增结果Fig. 1 Electrophoresis result of single PCR泳道M :DL 2000 DNA分子量标准 ;泳道1 :茄链格孢菌 ;泳道3 :立枯丝核菌(AG-3融合群);泳道5:致病疫霉 ;泳道2、4、6:阴性对照。Lane M: DL 2000 DNA marker; Lane 1: Alternaria solani; Lane 3:Rhizoctonia solani (AG-3); Lane 5: Phytophthora infestans; Lane 2, 4, 6:Negative control.
图2为单一PCR退火温度优化结果。由图2所示,退火温度为54℃时,致病疫霉的PCR产物条带最亮;退火温度为52℃时,立枯丝核菌(AG-3融合群)的产物条带最亮;而5个退火温度下,茄链格孢菌的PCR产物条带亮度差别不大(图2)。因此,本研究选择53℃作为多重PCR体系的退火温度。
2.2 多重PCR结果
2.2.1 引物浓度优化结果
由多重PCR 引物比例优化的结果可知(图3),当致病疫霉、茄链格孢菌和立枯丝核菌(AG-3融合群)的引物浓度分别为200、240和240 nmol/L时,3个条带都比较亮且亮度差别不大,因此该引物比例被选择作为最佳比例进行后续的试验。
2.2.2 特异性验证结果
当加入1、2或3种本研究所要检测的病原菌DNA时,PCR产物的电泳结果相应地也只出现加入菌的目的条带(图4:1~7泳道),而加入其他菌的DNA模板的多重PCR产物的电泳结果都没有条带(图4:8~13泳道),表明本研究所建立的多重PCR检测方法具有良好的特异性。

图2 单一PCR退火温度优化结果Fig. 2 Optimizations results of annealing temperature by single PCR(a)致病疫霉;(b)立枯丝核菌(AG-3融合群);(c)茄链格孢菌。泳道M: DL 2000 DNA分子量标准 ;泳道1~5 :退火温度依次为50、52、54、56、58℃ ;泳道C :阴性对照。(a) Phytophthora infestans; (b) Rhizoctonia solani (AG-3);(c) Alternaria solani. Lane M: DL2000 DNA marker;Lanes 1~5: Annealing temperature were 50, 52, 54, 56, 58 ℃,respectively; Lane C: Negative control.
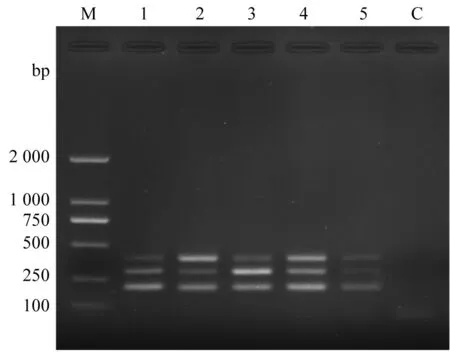
图3 多重PCR引物浓度优化结果Fig. 3 Optimizations result of primer concentration by multiplex PCR泳道M :DL 2000 DNA分子量标准 ;泳道1~5 :三对引物Pi-F/R、Rs-F/R和As-F/R的浓度分别为160、160和240 nmol/L,200、160 和240 nmol/L,160、240和200 nmol/L,200、240和240 nmol/L,160、160和160 nmol/L;泳道C:阴性对照。每对正反向引物的浓度相同。Lane M: DL 2000 DNA marker; Lanes 1~5: The concentrations of primer pair Pi-F/R, Rs-F/R and As-F/R were 160, 160 and 240 nmol/L,200, 160 and 240 nmol/L, 160, 240 and 200 nmol/L, 200, 240 and 240 nmol/L, 160, 160 and 160 nmol/L, respectively; Lane C: Negative control. The concentrations of forward primer and reverse primer in each pair are the same.
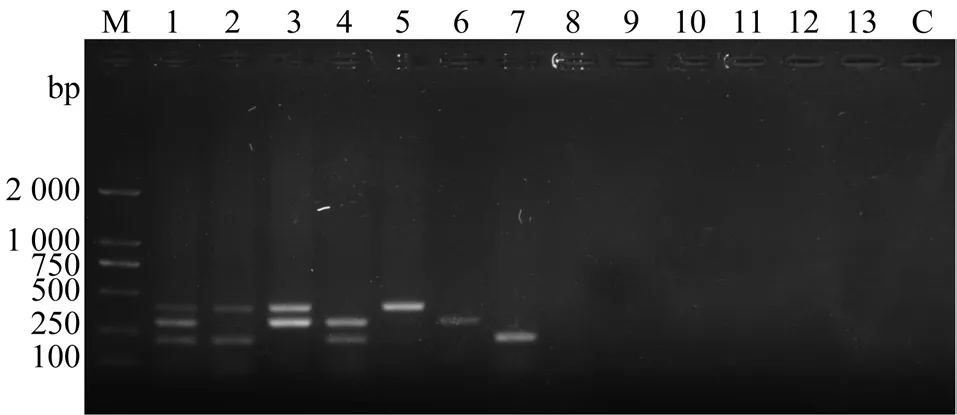
图4 多重PCR检测致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌的特异性结果Fig. 4 Specificity evaluation of the multiplex PCR for detection ofPhytophthora infestans, Rhizoctonia solani (AG-3) and Alternaria solani泳道M:DL 2000 DNA分子量标准;泳道1:致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌;泳道2:致病疫霉、茄链格孢菌;泳道3:致病疫霉、立枯丝核菌(AG-3融合群);泳道4:茄链格孢菌、立枯丝核菌(AG-3融合群);泳道5:致病疫霉;泳道6:立枯丝核菌(AG-3融合群);泳道7:茄链格孢菌;泳道8:大豆疫霉;泳道9:辣椒疫霉;泳道10:链格孢菌;泳道11:细极链格孢菌;泳道12:立枯丝核菌(AG-1融合群);泳道13:立枯丝核菌(AG-4融合群);泳道C:阴性对照。Lane M: DL 2000 DNA marker; Lane 1: P. infestans, R. solani (AG-3)and A. solani; Lane 2: P. infestans and A. solani; Lane 3: P. infestans and R. solani (AG-3); Lane 4: A. solani and R. solani (AG-3); Lane 5: P.infestans; Lane 6: R. solani (AG-3); Lane 7: A. solani; Lane 8: P. sojae;Lane 9: P. capsici; Lane 10: A. alternata; Lane 11: A. tenuissima; Lane 12:R. solani (AG-1); Lane 13: R. solani (AG-4); Lane C: Negative control.
2.2.3 灵敏度测定结果
如图5所示,在多重PCR体系中,当致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌的DNA质量浓度在40.0 pg/μL、4.0 pg/μL、400.0 fg/μL和40.0 fg/μL时,均有出现3条目的条带,且条带亮度随质量浓度降低逐渐减弱。当DNA的质量浓度降到4.0 fg/μL时,没有出现扩增条带。因此,该多重PCR方法对致病疫霉、茄链格孢菌和立枯丝核菌(AG-3融合群)的检测限均为40.0 fg/μL,且灵敏度较高。
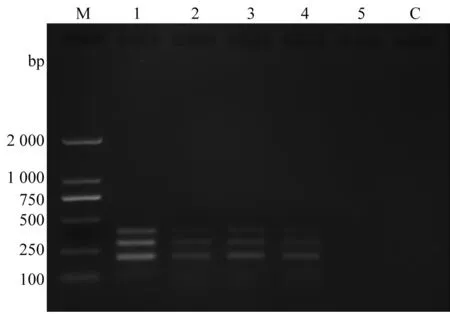
图5 多重PCR的灵敏度试验结果Fig. 5 Sensitivity assay of the multiplex PCR detection method泳道M :DL 2000 DNA分子量标准 ;泳道1~5 :多重PCR体系中模板DNA的质量浓度依次为40.0 pg/μL、4.0 pg/μL、400.0 fg/μL、40.0 fg/μL、4.0 fg/μL ;泳道C :阴性对照。Lane M: DL 2000 DNA marker; Lanes 1~5: DNA concentration in multiplex PCR were 40.0 pg/μL, 4.0 pg/μL, 400.0 fg/μL, 40.0 fg/μL,4.0 fg/μL, respectively; Lane C: Negative control.
2.2.4 土壤样品检测结果
土壤样品检测结果如图6所示,人工侵染的阳性土壤样品中均检测到3种菌所对应的条带,阴性样品和空白对照中均没有条带,表明本研究建立的多重PCR方法可应用于田间样品检测。
3 讨论
病害问题一直是制约马铃薯生产发展的主要因素。建立快速、准确、灵敏的病原菌检测方法对于马铃薯病害的预防和控制具有重要意义。传统的病原菌检测(如症状观察、分离纯化、形态学鉴定等手段)操作较复杂且时效性差,有的病害还极容易与其他病害相混淆,难以准确鉴定[19-20]。此外,田间的不少病害是由多种病原菌混合侵染导致的,这增加了检测的复杂度,传统的检测方法不能满足实际的生产需求[21]。分子生物学检测方法具有灵敏、特异和快速等优点,因能够克服传统检测方法的缺点而被广泛应用于病害鉴定和病原菌检测中[22]。
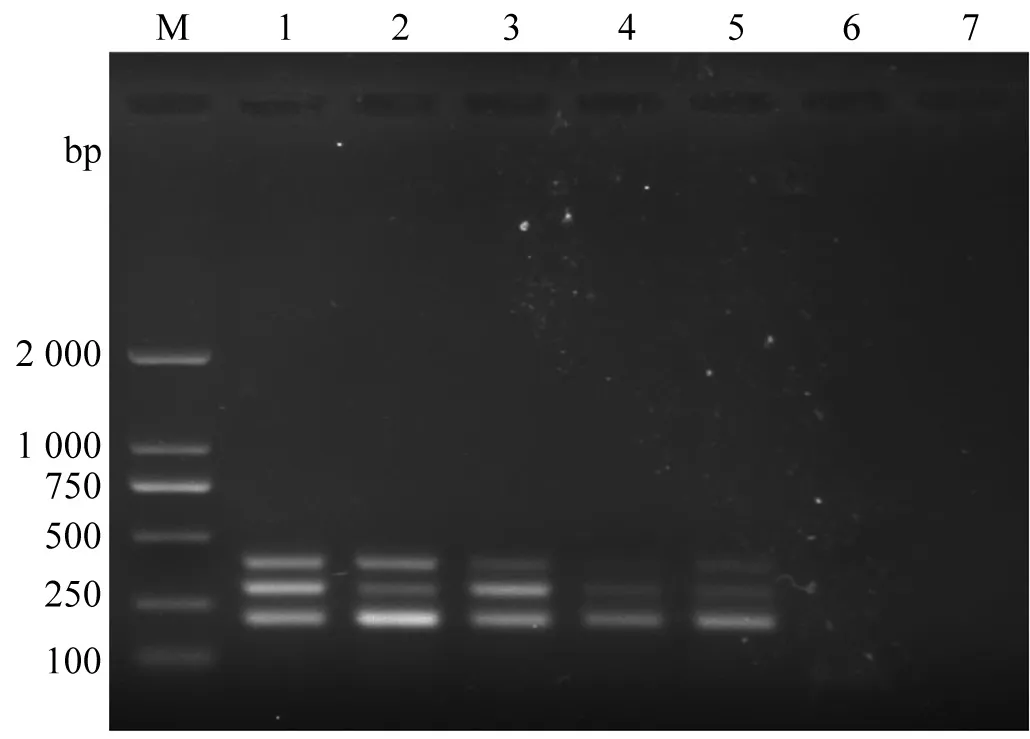
图6 土壤样品检测结果Fig. 6 Detection results of soil samples by multiplex PCR泳道M :DL 2000 DNA分子量标准 ;泳道1~5 :阳性土壤样品 ;泳道6:阴性土壤样品;泳道7:空白对照。Lane M: DL 2000 DNA marker; Lanes 1~5: Positive soil samples;Lane 6: Negative soil sample; Lane 7: Blank control.
多重PCR技术因其一个反应就能同时检测多种病原菌而受到许多研究者的青睐。国内外关于利用多重PCR检测马铃薯病原的研究多集中于病毒的分类或鉴定[23-24]。罗文彬等[25]采用多重RT-PCR方法建立了同时检测马铃薯A病毒(Potato virus A, PVA)、马铃薯M病毒(Potato virus M, PVM)、马铃薯S病毒(Potato virus S, PVS)、马铃薯 X 病毒(Potato virus X,PVX)和PVY的方法。Chikh Ali等[26]通过多重PCR不仅能鉴定出已知的PVY株系,还能鉴定新的重组株系。此外,陈庆河等[27]将双重PCR应用于致病疫霉(P. infestans)和青枯病菌(Ralstonia solanacearum)的快速检测中。多重PCR技术具有较高的灵敏度,能提高检测效率,降低试验成本。
马铃薯晚疫病、早疫病和黑痣病传染性强,均是严重影响马铃薯产业发展的病害。在田间,这3种病害有可能同时发生。因此,本研究针对这3种病害的病原菌[即致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌]成功地建立了一种同时检测这3种病原菌的多重PCR方法。该方法对致病疫霉、立枯丝核菌(AG-3融合群)和茄链格孢菌扩增出的目标片段大小分别为401、307和209 bp,凝胶电泳结果容易区分辨别,检测限为40.0 fg/μL,显示出了较高的灵敏度,且具有良好的特异性。综上所述,我们所建立的多重PCR检测方法具有很好的可行性,能够为马铃薯晚疫病、早疫病和黑痣病的早期预防、诊断及防控提供技术支持。
猜你喜欢
浙江农林大学学报(2022年5期)2022-10-12
绵阳师范学院学报(2021年5期)2021-05-28
数学大王·低年级(2020年8期)2020-08-14
河北大学学报(自然科学版)(2020年1期)2020-01-15
陕西农业科学(2019年2期)2019-04-12
生物工程学报(2019年1期)2019-01-30
现代食品(2018年5期)2018-06-06
中国酿造(2017年11期)2017-12-06
广东农业科学(2017年5期)2017-08-29
小雪花·初中高分作文(2016年9期)2016-05-14
