
桃叶珊瑚苷对Aβ25-35诱导的突起萎缩和神经元凋亡的保护作用
2020-10-28 09:54马智慧杨志友
中国药理学通报 2020年10期
马智慧,杨志友
(广东海洋大学食品科技学院海洋药物研究所,广东 湛江 524088)
阿尔茨海默病(Alzheimer’s disease,AD)是一种进行性的神经退行性疾病,发病原因复杂,其中β-淀粉样蛋白(Amyloid β,Aβ)是最为经典的致病因素之一[1]。目前为止,还没有任何药物能够阻止或逆转中重度AD病人记忆衰退的进程。神经突起构成的神经环路和神经网络对记忆的产生、巩固和存储起到主要作用[2-3]。有报道显示,Aβ诱导的神经突起萎缩和突触丢失构成了AD的病理学进程,启动并加速了神经元的凋亡,这些都将促进神经网络的破坏,最终导致记忆丢失[4-5]。因此,我们假设通过神经突起再生和神经元的保护,从而重建受损的神经网络可能是AD记忆修复的关键。我们前期大量的研究也证明,通过神经突起(轴突和树突)的再生而修复受损的神经网络与AD的记忆修复存在很大的相关性[6-8]。
传统药物为神经退行性疾病的治疗提供了更多的选择性,是AD候选药物开发的重要源泉,我们也从传统中药中发现了一些有助于改善记忆的有效成分[7-8]。桃叶珊瑚苷(Aucubin,AUC)作为一种环烯醚萜苷类化合物具有广泛的药理学活性,如抗炎、抗氧化、抗肿瘤、神经保护等[9-10]。然而,AUC是否具有改善Aβ诱导的神经元损伤的作用未见报道。因此,本研究从AUC抗神经损伤的角度出发,探讨其对Aβ诱导的神经元保护和突起再生的作用,为其治疗AD 提供理论和实验依据。
1 材料与方法
1.1 材料
1.1.1细胞株 人神经母细胞瘤细胞(SH-SY5Y),购自中国科学院上海分院细胞库。
1.1.2药物与试剂 桃叶珊瑚苷(纯度99.9%,批号:DST180612-004),购自成都德思特生物技术有限公司;Aβ25-35(Sigma公司,批号:A4559);0.05%和0.25% Trypsin-EDTA(批号:25300-054和25200-072)、马血清(批号:26050-088)、胎牛血清(批号:10270-106)、神经基础培养基(批号:21103-049)、MEM培养基(批号:C11095500BT)、F12培养基(批号:C11765500BT)、B27(批号:17504044)、脱氧核糖核酸酶DNase I(批号:18047-019)、胰蛋白酶Trypsine抑制剂 (批号:17075-011)、CMF-HBSS(批号:14185-052),均购自Gibco公司;反式维甲酸RA(Solarbio公司,批号:A9120);CCK8(APExBIO公司,批号:K1018);抗体β3-tubulin(Santa Cruz公司,批号:sc-80005);DAPI(Biomol公司,批号:ABD-17507);Alex Fluor 594山羊抗鼠二次抗体(Abcam公司,批号:ab150116)。
1.1.3仪器 CO2培养箱(Crystal公司);高速冷冻离心机(Beckman公司);全自动酶标仪(Biotech公司);自动细胞计数仪(Biotech公司);倒置荧光显微镜(OLYMPUS公司)。
1.2 方法
1.2.1原代皮层神经元及SH-SY5Y细胞培养 动物实验和操作方法遵照广东海洋大学动物伦理委员会章程(SYXK2014-0053)。从胎鼠大脑皮层中分离神经元的方法参照之前的报道[8-9],简言之,取胚胎14 d的胎鼠(E14,ICR)大脑皮层,去掉硬脑膜、剪碎,加入0.05%的Trypsin-EDTA于37 ℃消化15 min,用含12%马血清、33.3 mmol·L-1葡萄糖和2 mmol·L-1L-谷氨酰胺的神经基础培养基(HS培养基)终止消化,然后加入6× 105U·L-1DNase I和0.3 g·L-1Trypsine抑制剂,37 ℃孵育15 min,加入HS培养基后,离心(90×g, 3 min)去上清,用CMF-HBSS缓冲液重悬、离心两次,将细胞重悬于HS培养基中,过0.45 μm滤膜,将细胞接种于8-孔板中用含2% B27、33.3 mmol·L-1葡萄糖和2 mmol·L-1L-谷氨酰胺的神经基础培养基(B27培养基)培养。SH-SY5Y细胞用含15% FBS 的MEM/F12培养基于37 ℃、5% CO2的饱和湿度培养箱中培养。
1.2.2CCK-8法检测细胞存活率
1.2.2.1 Aβ25-35致SH-SY5Y损伤模型的建立及AUC的作用 实验分为正常对照组、模型组(Aβ25-3520 μmol·L-1)、AUC组(0.01、0.1、1、10、25 μmol·L-1)。将SH-SY5Y细胞以1.56×104·cm-2的密度接种于96孔板(每组5个复孔)中,培养24 h,用全反式维甲酸(RA)诱导神经分化6 d后,加入不同浓度的AUC,2 h 后加入Aβ25-35,37 ℃培养24 h后,CCK-8法检测细胞存活率。
1.2.2.2 Aβ25-35致原代皮层神经元损伤模型的建立 将原代皮层神经元以6.25×104·cm-2的密度接种于96孔板(每组5个复孔)中,细胞培养3 d后,加入不同浓度的Aβ25-35(终浓度10、15、20 μmol·L-1)继续培养3 d。
1.2.2.3 不同浓度AUC对Aβ25-35致原代大脑皮层神经元损伤的影响 实验分为正常对照组、模型组(Aβ25-3510 μmol·L-1)、AUC组(1、10 μmol·L-1)。细胞培养3 d后,加入Aβ25-35和AUC处理3 d,用CCK-8法测定细胞存活率,每组5个复孔。
1.2.3免疫细胞化学法检测神经突起密度
1.2.3.1 Aβ25-35致原代皮层神经元突起萎缩模型的建立 将原代皮层神经元以2.86×104·cm-2的密度接种于8孔板中,细胞培养3 d后,加入10 μmol·L-1的Aβ25-35继续培养3~4 d。
1.2.3.2 不同浓度AUC对Aβ25-35致皮层神经突起萎缩的影响 实验分为正常对照组、模型组(Aβ25-3510 μmol·L-1)、AUC组(1、10 μmol·L-1)。细胞培养3 d后,同时加入Aβ25-35(10 μmol·L-1) 和不同浓度的AUC (1、10 μmol·L-1),培养4 d后,弃去上清,用4% PFA固定1 h,加入一抗β3-tubulin (1 ∶ 500)于4 ℃过夜孵育,然后加入荧光594标记的山羊抗鼠二抗(1 ∶ 300),DAPI(1 mg·L-1)用于细胞核的染色。使用倒置荧光显微镜拍摄259 μm×346 μm大小的照片,每组拍摄10张,用ImageJ (NIH)软件分析β3-tubulin染色阳性的神经突起密度。
1.2.3.3 不同浓度AUC对Aβ25-35致皮层神经突起萎缩后再生的影响 实验分为正常对照组、模型组(Aβ25-3510 μmol·L-1)、AUC组(1、10 μmol·L-1)。原代皮层神经元培养3 d后,加入Aβ25-35(10 μmol·L-1)培养3 d,然后移去Aβ25-35,加入AUC继续培养4 d后,用如上方法对神经元进行染色并统计神经突起的密度。
1.2.3.4 不同浓度AUC对正常皮层神经突起再伸长的影响 实验分为正常对照组、AUC组(1、10 μmol·L-1)。取原代皮层神经元以2.14×104·cm-2的密度接种于8孔板中,培养2 d后,加入AUC继续培养4 d,用如上方法对神经元进行染色并统计神经突起的密度。
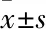
2 结果
2.1 AUC对Aβ25-35致SH-SY5Y细胞损伤的保护作用本课题组之前的研究结果发现Aβ25-35(20 μmol·L-1)作用于SH-SY5Y细胞24 h后,与对照组相比有明显的细胞损伤[11],因此选择20 μmol·L-1的Aβ25-35建立AD体外细胞系损伤模型(Fig 1A)。有文献报道显示0.01~10 μmol·L-1的AUC能明显促进神经前体细胞向GABA能神经元分化[12],因此我们设定AUC的浓度为0.01 ~ 25 μmol·L-1。系列浓度(0.01~25 μmol·L-1)的AUC作用于细胞2 h后,加入Aβ25-35处理24 h,与模型组相比,10和25 μmol·L-1的AUC组细胞存活率明显升高(P<0.01),表明AUC可以保护Aβ25-35诱导的SH-SY5Y细胞损伤(Fig 1B)。
2.2 AUC对Aβ25-35致小鼠原代皮层神经元损伤的保护作用为了验证AUC对神经元的保护作用,我们建立了胎鼠(孕期14 d)大脑皮层原代培养的神经元Aβ诱导损伤模型,Fig 2A的结果显示,10~20 μmol·L-1的Aβ25-35作用于神经元3 d后,细胞存活率明显降低,与对照组相比差异有显著性(P<0.01),因此我们选择10 μmol·L-1的Aβ25-35建立原代神经元体外AD模型。我们选取了AUC在SH-SY5Y细胞上有效浓度临界值的两个点(1和10 μmol·L-1)进行验证和后续实验。AUC与Aβ25-35共培养3 d后,与模型组相比,1和10 μmol·L-1的AUC组细胞存活率明显升高(P<0.05),表明AUC可以保护Aβ25-35诱导的原代神经元损伤(Fig 2B)。
2.3 AUC对Aβ25-35致小鼠原代皮层神经元突起萎缩的再生作用神经元轴突和树突构建的大脑神经环路和神经网络,对记忆的存储和回溯起到至关重要的作用[2-3],因此我们构建了神经突起萎缩模型,探讨AUC对神经突起萎缩的再生和保护作用(Fig 3A)。结果显示,10 μmol·L-1的Aβ25-35作用于神经元4 d后,与对照组相比,β3-tubulin阳性的突起密度明显降低(P<0.01),而AUC与Aβ25-35共培养干预4 d后,与模型组相比,10 μmol·L-1的AUC组突起密度明显升高(P<0.05),表明AUC可以保护Aβ25-35诱导的原代神经元突起萎缩(Fig 3A, C)。
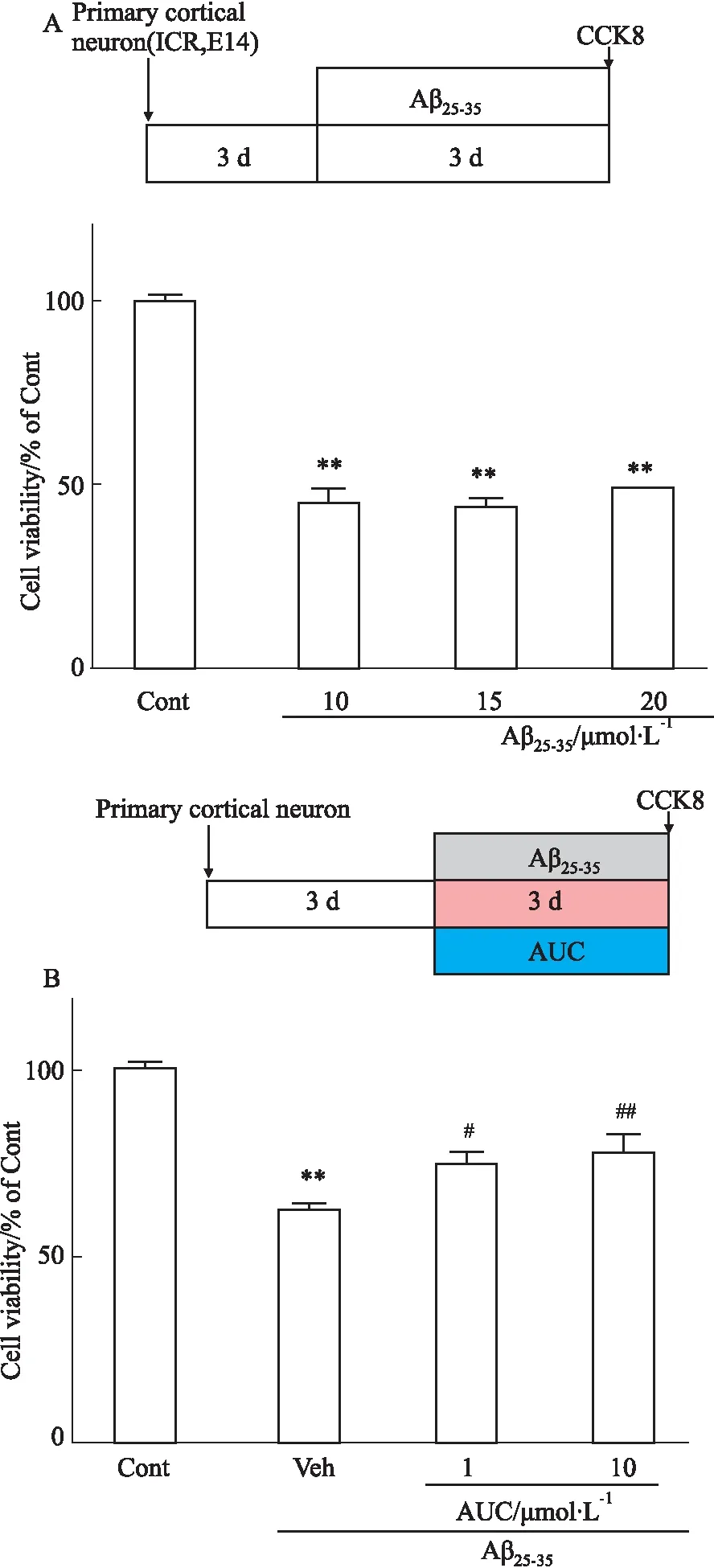
Fig 2 Protective effect of AUC on Aβ25-35-induced neuronal apoptosis in primary cultured cortical
为了进一步验证AUC对Aβ诱导的神经突起萎缩的再生作用,Aβ作用于原代培养的神经元3 d后,加入AUC培养4 d(Fig 3B)。结果显示,模型组与对照组相比,β3-tubulin阳性的突起密度明显降低(P<0.01);与模型组相比,10 μmol·L-1的 AUC处理后, 神经突起的密度明显增高(P<0.01)。表明AUC可以促进Aβ25-35诱导的原代神经元萎缩突起的再生(Fig 3B)。
2.4 AUC对正常原代皮层神经元突起再伸长的作用接下来我们探讨了AUC对正常培养的神经元轴突再生的作用(Fig 4A),免疫荧光染色结果表明,与对照组相比,AUC 1和10 μmol·L-1给药组能明显增加β3-tubulin阳性染色的突起密度(P<0.05)。进一步证明AUC具有促进神经元突起再生的作用(Fig 4B, C)。
3 讨论
Aβ老人斑和神经元纤维缠结是AD的两大病理性特征,迄今为止,β-淀粉样蛋白假说仍普遍被认为是AD的发病机制,有研究报道显示,Aβ会升高糖原合成酶激酶3β(GSK3β)和细胞周期素依赖蛋白激酶5(CDK5)的磷酸化水平,进而促进脑衰反应调节蛋白2(CRMP-2)的磷酸化,加速微管蛋白解聚使神经突起萎缩,并导致神经元的凋亡[4-6]。近年来,以Aβ或Tau蛋白为作用靶点的药物均以失败告终,而有一些天然药物能够促进神经突起再生但未表现出对Aβ降解作用却能明显改善记忆[13],这也提示我们仅仅抑制AD的病理因素不足以逆转其进程。因此,我们希望找到一些药物能够通过保护神经元并促进萎缩的神经突起再生的方式重新建立新的神经网络,达到改善和恢复AD记忆的作用。

Fig 3 Neurite regenerative effect of AUC on Aβ25-35-induced neurite atrophy in primary cultured cortical

Fig 4 Neurite regenerative effect of AUC on normal cultured
SH-SY5Y和PC12细胞被广泛应用于活性化合物筛选的体外细胞实验[14-15],通常分化成熟的神经元需要多次维甲酸(RA)诱导形成,故本实验采用RA诱导分化成熟的SH-SY5Y细胞用于AD有效化合物的体外筛选研究,并通过构建能够反映体内生理状态的原代大脑皮层神经元体系,进一步评价药物对神经元的保护和突起再生作用。本实验用Aβ25-35诱导SH-SY5Y细胞和神经元后,CCK8法检测细胞存活率,结果显示,与模型组相比,AUC组能够明显提高神经元存活率,表明其具有很好的神经保护作用。有报道也表明,AUC具有保护糖尿病脑病大鼠海马CA1区神经元损伤的作用[16]。体外细胞研究显示,AUC对H2O2诱导的PC12细胞的保护作用可能是通过抑制caspase激活实现的[17]。
鉴于Aβ诱导的AD发病机制和药物的改善作用机制可能是通过不同的通路实现的,因此我们在构建AD的药物药效评价模型时选取了不同的药物处理模式。 Aβ加入前进行药物预处理是先激活某一信号通路构筑细胞的防御体系;Aβ与药物同时加入可能是抑制了Aβ的信号通路或者通过激活其他信号通路拮抗了Aβ的作用,还可能是药物对Aβ寡聚体的分解作用;Aβ加入后进行药物处理主要是评价药物对细胞功能的再修复作用。本文建立了AUC在不同阶段对SH-SY5Y细胞和皮层原代神经元的干预模型,揭示了其对神经元的保护和突起再修复的作用。
神经轴突和树突的再生是重新建立和修复受损神经网络从而改善记忆的一种重要方式,我们前期的研究结果表明,促进神经元轴突再伸长与改善AD记忆功能之间存在很好的相关性[6],而促进体外神经元轴突再伸长的化合物能够明显增强正常小鼠的认知和空间记忆能力,临床研究也进一步证明了该类化合物具有明显提高健康人记忆的作用[18]。本研究发现,AUC能够保护并促进Aβ25-35诱导萎缩的神经元突起再生,且促进正常神经元突起的伸长。有研究表明,AUC能够促进神经干细胞和坐骨神经轴突再伸长,并促进神经元前体细胞向GABA氨基丁酸能神经元分化,具体作用机制尚待进一步的研究[12]。综上所述,通过建立AD体外细胞损伤模型,我们首次发现AUC对Aβ25-35诱导的原代皮层神经元的损伤具有保护作用,且对其诱导的神经突起萎缩具有再生作用。本研究为后续在体研究AUC是否具有改善记忆作用奠定了基础,以期更好的阐明其在神经退行性疾病中的应用价值。
猜你喜欢
贵州医科大学学报(2022年11期)2022-12-23
航天电子对抗(2022年4期)2022-10-24
中国应用生理学杂志(2022年2期)2022-08-29
中国医学影像技术(2020年11期)2021-01-04
山西大同大学学报(自然科学版)(2020年3期)2020-07-17
中国现代医药杂志(2020年3期)2020-05-08
遵义医科大学学报(2019年4期)2019-10-22
中国科技纵横(2018年2期)2018-11-29
中国当代医药(2017年17期)2017-07-25
支部建设(2016年18期)2016-11-28
