
FoxG1-亨廷顿病潜在的靶向调控因子
2020-10-28 08:42马思飞朱永强胡沿每宋黄戎郭美娜张维宁刘子重钱进军
中国药理学通报 2020年10期
马思飞,恽 琪,朱永强,2,胡沿每,宋黄戎,郭美娜,张维宁,刘子重,2,钱进军,2,王 佳,2
(江苏大学 1.医学院,江苏 镇江 212013;2. 第四人民医院神经内科、介入科,江苏 镇江 212001)
亨廷顿舞蹈症(Huntington’s disease,HD)是一种迟发性(发病年龄一般为30~50岁)的神经退行性病,主要表现为运动、认知以及记忆功能的丧失,患者发病后15~20年死亡。该病具有高度的脑区选择性,主要侵害基底神经节和大脑皮层,表现为纹状体投射性GABA神经元和大脑运动皮层锥体神经元死亡[1]。基底神经节纹状体受损引起HD典型的运动过度-舞蹈样动作;大脑皮层受损诱发患者认知和情感障碍,晚期患者多见痴呆。1993年,HD课题组通过外显子扩增和cDNA克隆技术将HD基因it-15定位于染色体4p16.3,该突变基因编码一分子量为350 ku的蛋白-亨廷顿蛋白(Huntingtin,Htt)。Htt由3144个氨基酸组成,氨基末端第17位氨基酸残基有一段多聚的谷氨酰胺(PolyQ)序列,其长度在正常人群中呈多态性,但均短于35个,在HD患者体内,突变的亨廷顿蛋白(Mutant Huntingtin,mutHtt)PolyQ序列的长度超过37个[2]。PolyQ重复拷贝数的异常扩增成为HD发病的基础。
HD发病的分子机制尚不清楚。越来越多的证据表明,转录调控异常与HD发生密切相关[3]。无论在HD细胞模型、转基因小鼠模型还是HD患者脑组织研究结果均证实mRNA转录水平的异常。突变的亨廷顿蛋白片段入核后直接或间接地与一些转录因子、转录辅激活因子如CBP,SP,核阻遏物N-CoR等作用导致细胞基因的转录失调[4]。HD患者及模型鼠脑组织可见大量mutHtt片段,同时伴随Htt mRNA转录水平下调。DNA芯片技术也已经证实mutHtt引起mRNA转录水平异常[5]。
FoxG1作为作为叉头转录因子家族中的一个成员,除了参与调控胚胎期端脑神经发生的进程,也广泛表达于出生后神经元,并伴随整个成年期[6]。FoxG1与有丝分裂后神经元的存活密切相关,FoxG1的神经元保护功能是由信号通路介导的[7]。Wortmannin选择抑制PI3K-Akt信号通路后,FoxG1将无法逆转低钾诱导的神经元的死亡[8]。
亨廷顿病的发生与转录调控密切相关,然而转录调控因子FoxG1与亨廷顿病的关系却鲜见报道。本研究以亨廷顿体内模型(R6/2小鼠)及体外模型(细胞系、皮层神经元过表达mut-Htt)为研究对象,通过Westernblot、qRT-PCR及IF技术检测亨廷顿病发生后FoxG1的转录及表达水平的改变,为寻找亨廷顿病的潜在调控靶点及探讨亨廷顿病的发病机制提供实验基础。
1 材料与方法
1.1 材料
1.1.1试剂与抗体 细胞培养基均购买自Invitrogen(Carlsbad, CA),化学试剂购买自Sigma-Aldrich(St.Louis, MO),原代神经元培养的培养基购买自Trevigen(Gaithersburg, MD),实验所需的抗体如下:FoxG1 (Abcam, catalog # ab18259), FLAG (Sigma-Aldrich, catalog #F1804), α-Tubulin (Sigma-Aldrich, catalog #T9026), GFP抗体Santa Cruz Biotechnology (小鼠-GFP catalog #sc-9996a 和兔-GFP catalog #sc-8334). HRP标记的二抗 (碧云天,中国)。免疫荧光二抗Dylight 594 (catalog #115-515-044)和Alexa Fluor 488 (catalog #111-545-144)购买自Immuno Research Laboratories (West Grove, PA).
1.1.2质粒 FoxG1-flag质粒、Q15-GFP、 Q138-GFP质粒均由爱必梦生物科技有限公司构建。
1.1.3细胞系 HEK293T人胚胎肾细胞(cat. # CRL-3216),N2A神经母细胞瘤细胞(cat # CCL-131)购买自ATCC (Manassas, VA),小鼠HT22细胞由江苏大学胡嘉波教授惠赠。
1.1.4实验动物 ♂ R6/2转基因小鼠B6CBA-Tg(HDexon1)62Gpb/1J杂合子(购买自Jackson lab,Bar Harbor, ME, catalog #002810)与B6CBAF1/J 背景的♀小鼠(购买自南京模式动物所, catalog #100011)交配,通过PCR鉴定获得多聚PolyQ序列的长度至120 ± 5的亨廷顿病模型鼠。
1.2 方法
1.2.1原代神经元分离、培养 Poly-L-Lysine(Trevigen Gaithersburg,MD)包被玻片;皮层神经元分离及培养,从妊娠d 16~18大鼠的子宫剥离小鼠,取其前额皮层至冷HBSS(Invitrogen,Carlsbad, CA)中。将其前额皮层置于0.25% Trypsin-EDTA(Life Technologies)37 ℃水浴锅中消化。消化时间为9 min,其中每3 min将离心管混匀一下。加入20 %小牛血清培养基终止消化。用Neuronal Basial Medium(其中加入B27 Lot: 1760190, Life Technologisies; 青霉素, L-谷氨酰胺Lot: 1760127, Life Technologisies; 丙酮酸钠 Lot: 1181802,Life Technologisies)清洗血清及裂解细胞,铺板,5 d后进行神经元转染。
1.2.2脂质体转染 当培养的细胞密度达到40%时开始用Lipofectamine 2000(Life Technologies)进行脂质体转染:管1:500 μL OMEM + 8 μg DNA;管2:500 μL OMEM+15 μL Lipo;混匀,静置5 min;管1+管2,混匀,静置25 min;加入HEK293T细胞中;次日,收细胞。转染细胞活力通过细胞免疫荧光或DAPI染色确定。
1.2.3磷酸钙转染 皮层神经元培养后d 6进行转染:管1: CaCl27.5 μL/4孔;DNA15 μL/4孔;水52.5 μL/4孔;管2: HBS 75 μL/4孔;保存4孔板中的条件性培养基至CO2孵箱中(ThermoFisher),加入Neuroal Basial Medium;管1+管2,等待5~7 min;加入4孔板中,CO2孵箱中孵育20~30 min;替换回保留的条件性培养基。
1.2.4Western blot 转染后24 h裂解、收集细胞,蛋白浓度定量,按照WB实验流程操作完成:培养细胞上样量为50 μg/孔,上样至10%凝胶中,SDS-PAGE电泳(Bio-Rad)(130 V,1.5 h),转膜(Bio-Rad)(35 V,过夜),5% 牛奶封闭1 h,TBST 洗3次,10 min/次,兔抗FoxG1单克隆一抗(1 ∶ 1 000),鼠抗α-Tubulin(1 ∶ 40 000),4 ℃孵育过夜,山羊抗兔IgG二抗(1 ∶ 5 000);山羊抗鼠IgG 二抗(1 ∶ 5 000)孵育1 h。4 ℃孵育过夜,蛋白表达量使用Image J分析软件进行统计。样本检测结果通过α-Tubulin校正。
1.2.5qRT-PCR ① RNA提取:转染后24 h收集、裂解细胞,加入1 mL TRIzol(Life)室温5 min,加入200 μL氯仿剧烈震荡15 s,静置2~3 min,离心10 000×g, 4 ℃,10 min,转移水相置于新的EP管,异丙醇还原RNA,室温静置10 min,离心10 000×g, 4 ℃,10 min,弃上清,体积分数为0.75的乙醇清洗沉淀,离心7 200×g,4 ℃, 5 min,静置5~10 min。
② RNA逆转录为cDNA:向RNA沉淀加入20 μL DEPC水,测试浓度后,按照试剂盒Verso cDNA synthesis kit(Thermo catalog # ab-1453/B)将RNA逆转录成cDNA。简述如下:3 μg RNA样品加入试剂盒提供的反应预混液(包括反转录酶,反转录引物混合物以及缓冲液),42 ℃水浴中培育60 min合成所需cDNA模板。最后95 ℃处理2 min使反转录酶失活。
③ qRT-PCR:荧光定量PCR(Thermo Fisher)将会按照SYBR Green试剂盒的用户说明书进行操作。简述如下:每个PCR管含有25 μL反应液:包括100 ng cDNA模板,1X SYBR Green PCR预混液,1 μmol·L-1的正反引物。FoxG1引物(247 bp)序列分别:5′-CTGGGCAACAACCACTCCTT-3′和5′-CTTCTTCA TCATCTCCCTGGTGATGTGCTGGTCTGCGAAGTCATT-3′。β-actin基因的mRNA将会用于内参对照,β-actin引物(721 bp)序列分别为:5′-TCATGAAGTGT GACGTTGACATCCGTAAAG-3′和5′-CCTAGAAGCA TTTGCGGTGCACGATGGAGG-3′。
1.2.6细胞免疫荧光 转染后d 2,进行细胞免疫荧光:4%多聚甲醛固定细胞; 0.2% TritonX-100孵育10 min;5% BSA+5% Goat Serum 室温孵育90 min;一抗4 ℃孵育过夜;二抗室温孵育45 min;DAPI 核染10 min;封片。
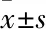
2 结果
2.1 离体细胞水平
2.1.1亨廷顿病伴随着FoxG1蛋白表达水平降低 ① N2A细胞系:以N2A细胞系为研究对象,脂质体法转染GFP、Q15-GFP、Q138-GFP质粒,Western blot检测各组FoxG1,GFP及Tubulin水平,Bonferroni's Multiple Comparison Test统计学结果显示,Q138诱导的亨廷顿病模型组FoxG1表达水平较空白对照组(GFP组)明显降低[F2,6=29,P=0.000 3],较正常对照组(Q15过表达组)也明显下降[F2,6=29,P=0.002 4],见Fig 1。
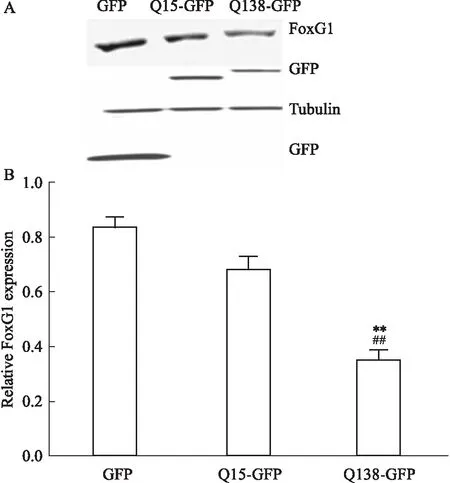
Fig 1 Expression level of FoxG1 in N2A cell line
② HT22细胞系:以HT22细胞系为研究对象,脂质体法转染PKL6.1、GFP、Q15-GFP、Q138-GFP质粒,WB检测各组FoxG1及Tubulin水平,Bonferroni's Multiple Comparison Test统计学结果显示,Q138诱导的亨廷顿病模型组FoxG1表达水平较正常对照组(Q15过表达组)明显下降[F3,8=38.58,P<0.05],见Fig 2。
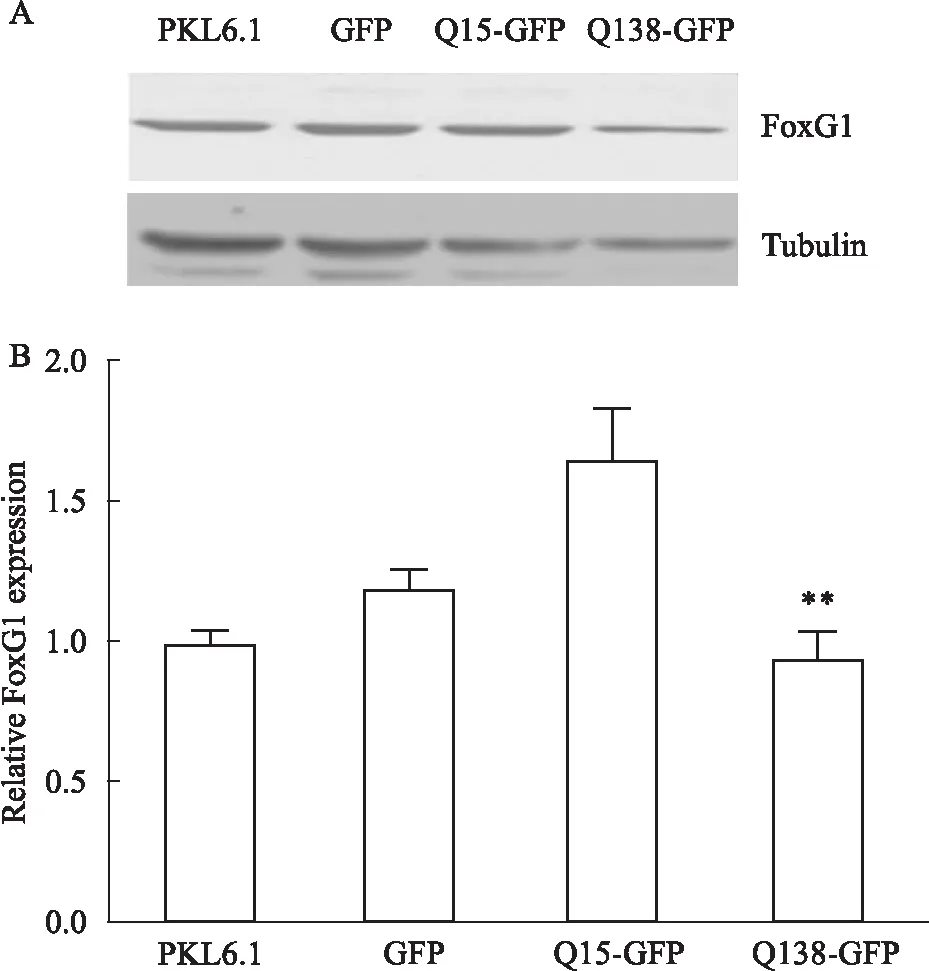
Fig 2 Expression level of FoxG1 in HT22 cell line
③ 皮层神经元:以皮层原代神经元为研究对象,磷酸钙法转染Q15-GFP,Q138-GFP质粒,WB检测FoxG1蛋白表达,Studentttest结果显示,Q138诱导的亨廷顿病模型组FoxG1表达水平明显下降[P=0.002],见Fig 3。
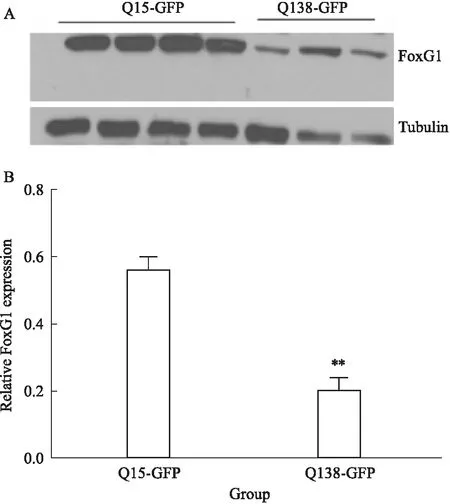
Fig 3 Expression level of FoxG1 in primary cortical neuron
2.1.2亨廷顿病伴随着FoxG1转录水平降低 N2A及HT22细胞系:以N2A及HT22细胞系为研究对象,脂质体法转染Q15-GFP和Q138-GFP质粒建立亨廷顿细胞模型,qRT-PCR检测各组FoxG1的转录水平,studentttest结果显示N2A细胞系[P=0.0417]及HT22细胞系[P=0.045 72]过表达Q138质粒均能明显下调FoxG1的转录水平,见Fig 4。
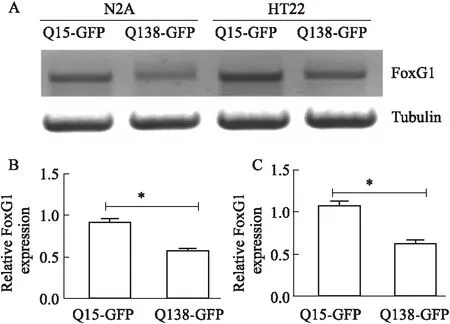
Fig 4 Transcription level of FoxG1 in N2A and HT22 cell lines
2.1.3FoxG1具有神经元的保护功能 皮层神经元 磷酸钙沉淀转染法在皮层神经元分别过表达GFP、Q138、FoxG1及Q138+FoxG1质粒,细胞免疫荧光检测各组细胞的状态(Fig 5),Q138质粒转染诱导了细胞核浓缩、聚集,FoxG1过表达组明显改善了Q138质粒诱导的神经元死亡,细胞核重新恢复至正常状态。
2.2 在体水平R6/2转基因小鼠 对比6周和12周亨廷顿模型(R6/2转基因小鼠)海马、前额皮层及纹状体FoxG1的蛋白表达,TWO-WAY ANOVA结果显示与对照组相比,R6/2转基因小鼠(6周)FoxG1蛋白表达在海马、前额皮层及纹状体均无显著差别;R6/2转基因小鼠(12周)前额皮层[F1,4=43.38,P<0.05]及纹状体[F1,4=38.5,P<0.05]FoxG1蛋白表达明显下降。
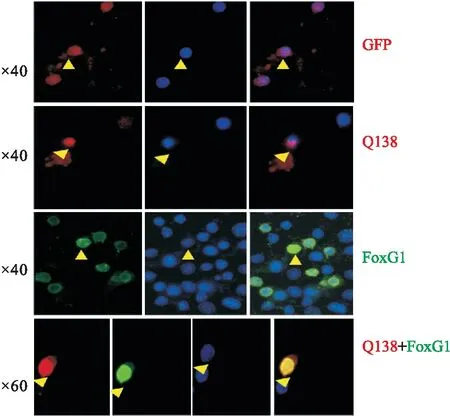
Fig 5 Protective function of FoxG1 for cortical neurons over expressed with mut-Htt
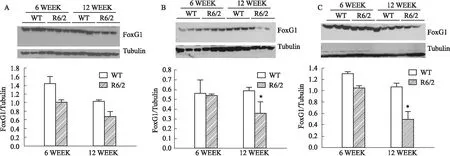
Fig 6 Expression level of FoxG1 in WT and R6/2 transgenic n=3)
3 讨论
越来越多的研究表明,许多神经退行性疾病:如亨廷顿病、帕金森氏综合症及阿尔茨海默病的发生均与转录调控因子(转录活化因子、转录抑制因子)的异常密切相关[9-10]。当前的研究通过体内、体外实验充分证明了神经退行性疾病-亨廷顿病的发生伴随着转录因子FoxG1转录及蛋白表达水平下降,FoxG1过表达明显改善了亨廷顿病所伴随的神经元的死亡。
本研究通过脂质体转染法及磷酸钙沉淀法将mutHtt(PolyQ序列的长度为138个)转染至N2A、HT22及皮层神经元,成功构建了HD的细胞模型,并发现HD的发生伴随转录调控因子-FoxG1转录及蛋白表达水平下降。
此外,该结果在体内实验再次得到验证:R6/2转基因小鼠(多聚PolyQ序列的长度至120 ± 5的亨廷顿病模型鼠)FoxG1蛋白表达水平明显下降,与海马脑区相比,前额皮层及纹状体FoxG1的蛋白表达水平显著降低。HD的发病具有高度的脑区选择性,主要侵害基底神经节,表现为纹状体投射性GABA神经元死亡,基底神经节纹状体受损引起HD典型的运动过度-舞蹈样动作,与当前的实验结果相一致[11]。
许多研究证实亨廷顿病的发生往往伴随着神经元的大量丢失[12],课题组通过过表达Q138质粒成功诱导了HD的细胞模型:IF显示HD模型组神经元细胞核皱缩、聚集,形状、亮度出现异常,FoxG1过表达显著逆转了HD细胞核的异质化,并恢复至GFP对照组水平,细胞形态、数目正常。
FoxG1与神经发育之间的关系已得到大量研究。然而,FoxG1对于有丝分裂后成熟神经元的功能却知之甚少。FoxG1除了参与调控神经细胞增殖、分化外,对于神经前体细胞的存活也起着非常重要的作用。2016年霍德华·休斯医学研究所通过Airyscan技术单细胞定位显微镜实时捕捉到了mutHtt与FoxG1家族蛋白的共聚体,验证了FoxG1通过自噬机制参与了mutHtt的清除,减少mutHtt诱导的细胞毒作用。当前的结果证实,FoxG1通过神经元的保护功能,改善HD所伴随的神经元死亡,但其潜在的分子生物学机制有待于进一步研究。
综上所述,亨廷顿病的发生伴随转录调控因子FoxG1转录及蛋白表达水平下降。FoxG1通过神经元的保护功能,改善亨廷顿病所伴随的神经元死亡,转录调控因子FoxG1为亨廷顿病的潜在调控位点,对亨廷顿病的机制研究及靶向药物研发具有重要价值。
(致谢:本课题是在江苏大学医学院行为医学实验室完成,感谢课题组肖栩、薛程、李晓晖、周漾同学参与了本课题的行为学实验。)
猜你喜欢
科学与生活(2021年16期)2021-11-25
中国医学影像技术(2020年11期)2021-01-04
浙江医学(2020年19期)2020-10-20
中国现代医药杂志(2020年3期)2020-05-08
科学24小时(2019年4期)2019-06-10
医药前沿(2019年29期)2019-01-05
中国当代医药(2017年17期)2017-07-25
支部建设(2016年18期)2016-11-28
科学生活(2016年7期)2016-07-25
医学研究杂志(2015年9期)2015-07-01
