
大肠杆菌过氧化物酶EfeB在细胞氧化应激中的作用
2020-09-23 09:21丁亮亮刘进生顾鹏帅唐蕾
食品与发酵工业 2020年17期
丁亮亮,刘进生,顾鹏帅,唐蕾,2*
1(工业生物技术教育部重点实验室(江南大学),江苏 无锡,214122)2(江南大学 生物工程学院,江苏 无锡,214122)
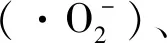
为了应对氧化应激,生物体已经进化出多种保护系统,其中对于ROS的清除一般包含非酶系统和酶系统。非酶系统通常是生物体自身合成的一类具有还原性的小分子物质,如谷胱甘肽、维生素C、海藻糖以及一些酮酚类物质等[7]。酶系统一般有超氧化物歧化酶、过氧化氢酶、过氧化物酶以及葡萄糖6-磷酸脱氢酶等[8-10],其中过氧化物酶属于氧化还原酶,广泛存在于各种生命体中,大多数是血红素过氧化物酶,并可以利用H2O2催化其他化合物,在生物合成、降解及防御等方面发挥重要作用[11]。过氧化物酶EfeB,最初被作为大肠杆菌“双精氨酸”易位系统的底物进行了研究,是第一个公认的细菌染料脱色过氧化物酶[12]。后续研究表明,EfeB是以血红素为辅基的过氧化物酶,而且具有特异性的铁转运功能[13]。目前对于染料脱色过氧化物酶的研究主要集中在提高酶活力及降解底物方面[14-16],大肠杆菌中关于EfeB在氧化应激条件下对胞内生理状况的调控机制尚不清楚。
本文以E.coliBL21(DE3)为出发菌株,通过efeB的敲除及过量表达,分别探究了在不同条件下efeB对菌体生长、丙二醛浓度、ROS合成以及抗氧化途径关键酶基因表达水平的影响,为寻找调控胞内氧化应激的策略提供理论参考。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
实验所用菌株和质粒见表1;本研究中所用引物由天霖生物科技(无锡)有限公司合成,引物详细信息见表2。

表1 本研究中使用的菌株与质粒Table 1 Strains and plasmids used in this study

表2 本研究所用引物Table 2 Primers used in this study
1.1.2 主要试剂与仪器
限制性内切酶EcoR I、XhoI和T4 DNA连接酶、质粒提取、纯化试剂盒,TaKaRa公司;ClonExpress®II One Step Cloning Kit、ClonExpress®MultiS One Step Cloning Kit,南京诺唯赞生物科技有限公司;活性氧测定试剂盒,南京建成生物工程研究所;异丙基-β-D-硫代吡喃半乳糖苷(isopropyl-β-D-thiogalactopyranoside,IPTG)、硫酸卡那霉素(kanamycin,Kan)、氨苄青霉素(ampicillin,Amp)和氯霉素(chloramphenicol,Chl),生工生物工程(上海)有限公司。
核酸电泳仪、凝胶成像系统, Bio-Rad 公司;可见光分光光度计,尤尼柯(上海)仪器有限公司;Enspire 多标记检测系统(酶标仪),珀金埃尔默有限公司;激光共聚焦显微镜,德国徕卡公司;流式细胞仪,美国BD公司;实时荧光定量PCR仪,美国应用生物系统公司。
1.1.3 培养基及培养条件
LB 培养基(g/L):胰蛋白胨10,酵母粉5,NaCl 10,固体LB培养基再加入2%的琼脂。抗性培养基添加的Amp、Kan和Chl终质量浓度分别为100、50、25 mg/L,IPTG终浓度0.2 mmol/L。
摇瓶发酵条件:250 mL摇瓶装液量50 mL,37 ℃、200 r/min摇床培养。
1.2 实验方法
1.2.1 质粒构建
以E.coliBL21(DE3)的基因组为模板,设计引物UefeBF/UefeBR通过PCR扩增得到efeB目的片段(1272 bp)。纯化后的片段与载体pET28a经过EcoR I和XhoI酶切后,通过T4 DNA连接酶进行连接,并将连接产物转化到E.coliJM109感受态中,在Kan抗性平板上筛选阳性转化子,进行双酶切验证和基因测序(天霖生物科技(无锡)有限公司),将序列正确的质粒定名为pEE。
1.2.2 大肠杆菌efeB的敲除
以E.coliBL21(DE3)的基因组为模板,设计目的基因efeB上下游各500 bp的同源序列的引物。分别用引物对UefeBF/UefeBR和DefeBF/DefeBR扩增出efeB上游的同源序列基因UefeB和下游的同源序列基因DefeB,并以pKD4质粒为模板设计引物kanF和kanR,扩增含有Kan抗性片段及其两端FRT基因kan。将经过EcoR I和XhoI酶切后的线性质粒pET22b与以上扩增出的3个基因片段利用ClonExpress®MultiS One Step Cloning Kit试剂盒进行连接,并将连接产物转化入感受态E.coliJM109中,在Kan和Amp双抗平板上筛选阳性转化子,进行验证,最终得到质粒pEUKD。
进一步以质粒pEUKD为模板,利用引物KefeBF/KefeBR进行PCR扩增。PCR反应体系(50 μL):模板1 μL,Primerstar Max DNA polymerase 25 μL,上下游引物各1 μL,ddH2O 22 μL。PCR反应条件:95 ℃ 5 min;94 ℃ 30 s;X℃ 15 s;72 ℃Ys;30个循环;72 ℃ 10 min (其中退火温度X根据不同基因片段的引物Tm值进行设置,延伸时间Y则根据片段长度按照每分钟扩增1 000个碱基的速度进行分配设置)。最终得到两端带有500 bpefeB基因上下游同源序列,中间为含FRT位点的Kan抗性基因的DNA打靶片段,用于敲除efeB基因。
大肠杆菌efeB的敲除具体过程为:将2502 bp左右的线性打靶片段电转入感受态E.coliBL21(DE3)/pKD46后,并迅速加入1 mL LB液体培养基,在37 ℃、200 r/min条件下振荡复苏12 h进行同源重组,之后均匀涂布在含有Kan抗性的固体LB平板上,筛选阳性重组菌。挑取阳性单菌落,以E.coliBL21(DE3)为对照,利用引物对JefeBF/ JefeBR进行菌落PCR扩增,验证条带大小正确后,再导入pCP20质粒,30 ℃振荡培养4 h后,涂布至Amp抗性平板上,于30 ℃过夜培养。以E.coliBL21(DE3)为对照,利用引物JefeBF/JefeBR进行菌落PCR扩增,验证条带大小和测序验证正确后,再通过42 ℃培养,消除胞内质粒,得到没有任何抗性的EcoΔefeB[19]。
1.2.3 培养方法
将各菌株于LB 液体培养基中在37 ℃,200 r/min条件下活化12 h后,按体积分数1%接种量转移至50 mL LB液体培养基中培养并加入相应的抗生素,需要添加诱导剂的菌株,在培养2 h后,加入终浓度0.2 mmol/L的IPTG进行诱导培养。
1.2.4 重组蛋白表达及SDS-PAGE检测
参照CHEN等[17]实验方法检测重组菌株蛋白表达情况。
1.2.5 外源添加H2O2浓度的确定
将E.ColiBL21(DE3)于LB液体培养基中37 ℃下过夜培养后,按体积分数1%接种量接种至50 mL LB液体培养基中,培养至OD600为0.3,然后在摇瓶中添加不同终浓度的H2O2:1、2、4、8和16 μmol/L。没有H2O2处理的为对照组,在样品培养2、4、6、8、10和12 h后,测量并记录OD600[20]。
1.2.6efeB基因敲除及过表达对大肠杆菌生长的影响
测定方法同1.2.5。
1.2.7 细胞膜脂质过氧化水平的测定
参考曾昕[21]的方法。利用脂质过氧化产物丙二醛(malondialdehyde, MDA)与硫代巴比妥酸(thiobarbituric acid, TBA)的显色反应来测定。在对数期收集细胞,并与TBA以1∶1的体积比混合,置于沸水浴中12 min后迅速转入冰水浴中冷却。在室温条件下,经12 000 r/min离心10 min,取200 μL上清液至96孔酶标板中,通过酶标仪分别测定其在450、532和600 nm波长处的吸光值。并通过公式(1)计算MDA的含量(nmol/g蛋白)。
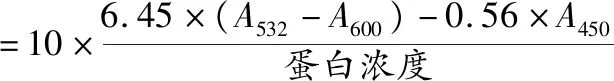
(1)
1.2.8 胞内活性氧检测
采用化学荧光法[22]。在菌体生长至对数生长期时,取200 μL菌液置于1.5 mL离心管中,加入800 μL 磷酸盐缓冲液进行稀释悬浮后,再加入浓度为10 mmol/L的2,7-二氯荧光黄双乙酸盐(2,7-dichlorofluorescent yellow diacetate, DCFH-DA)1 μL,充分混匀后置于37 ℃,200 r/min摇床中,反应1 h。12 000 r/min离心5 min弃上清液,收集菌体,用磷酸缓冲液洗涤2次后弃上清液,再加入200 μL的磷酸盐缓冲液重悬菌体并制片,在激光共聚焦显微镜下进行胞内ROS定性检测。其中,显微镜检测条件为:激发波长502 nm,发射波长530 nm。之后采用同样方法染色,取1 mL重悬菌液,利用流式细胞仪进行胞内ROS定量检测。
1.2.9 基因转录水平检测
efeB过表达菌株于37 ℃,200 r/min过夜培养后,以体积分数1%的接种量转接至新鲜LB培养基中,培养2 h后加入IPTG诱导剂,并诱导到对数生长期。在4 ℃,12 000 r/min离心5 min后收集菌体,并用50 mg/mL的溶菌酶在37 ℃下破壁10 min。按照试剂盒说明书进行总RNA提取,提取的RNA浓度及纯度使用核酸定量仪测定,之后将 RNA 直接反转录成cDNA用于荧光定量实验。以上述cDNA为模板进行荧光定量PCR,荧光定量PCR引物见表3,选择编码 3-磷酸甘油醛脱氢酶的基因gapA作为内参基因,采用SYBR®Green I嵌合荧光法,反应体系参照试剂盒说明书。

表3 本研究所用荧光定量 PCR 引物Table 3 Primers for fluorescent quantitative PCR used in this study
2 结果与分析
2.1 EcoΔefeB和Eco/pEE菌株的构建
以质粒pEUKD为模板,利用引物对KefeBF/KefeBR扩增得到长度为2 502 bp的打靶片段(图1-a)。将打靶片段电转进E.coliBL21(DE3)/pKD46 后,利用引物JefeBF/JefeBR进行菌落 PCR鉴定,同源重组成功后理论上可获得2 613 bp片段,而E.coliBL21(DE3)则为2 383 bp片段,结果表明挑取的菌落均可扩增出2 613 bp片段(图1-b),同源重组成功。转入pCP20质粒后,利用引物对JefeBF/JefeBR进行菌落pCR鉴定,efeB敲除成功的菌落理论上PCR结果为1 159 bp (保留一个FRT位点),而E.coliBL21(DE3) PCR结果为2 383 bp,结果表明挑取的菌落均可扩增出1 159 bp片段(图1-c),对验证正确的敲除菌测序,结果正确,表明efeB被成功敲除,并将敲除菌命名为EcoΔefeB。
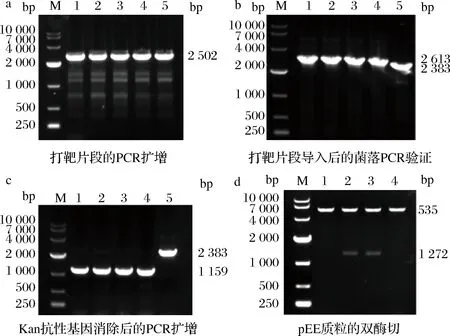
a-打靶片段的PCR扩增:1~5-打靶片段;b-打靶片段导入后的菌落PCR验证:M-Marker;1~4-打靶片段重组成功的菌株,5-E. coli BL21(DE3)对照;c-Kan抗性基因消除后的PCR扩增:M-Marker,1~4:efeB敲除菌株,5-E. coli BL21(DE3)对照;d-pEE质粒的双酶切:M-Marker,2~3-pEE图1 efeB基因敲除与重组质粒pEE的电泳验证Fig.1 Electrophoretic verification of efeB gene knockout and recombinant plasmid pEE
通过EcoRI和XhoI双酶切鉴定pEE质粒,efeB长度为1 272 bp,pET28a 质粒双酶切后为5 335 bp,双酶切条带大小与理论一致(图1-d),对构建的质粒测序,结果正确,将pEE转化入E.coliBL21(DE3)中,得到菌株Eco/pEE。
2.2 重组蛋白表达及SDS-PAGE检测
为了检测过表达菌株EfeB诱导后是否表达,对Eco/pEE菌株诱导后进行SDS-PAGE检测,EfeB蛋白分子量约为46 kDa,其蛋白表达检测结果如图2所示,过表达菌株在添加IPTG诱导后与对照菌株相比,检测到重组蛋白,目的条带的大小与电泳图中的位置吻合,因此EfeB蛋白表达量增加,重组菌株Eco/pEE可以用于后续实验。
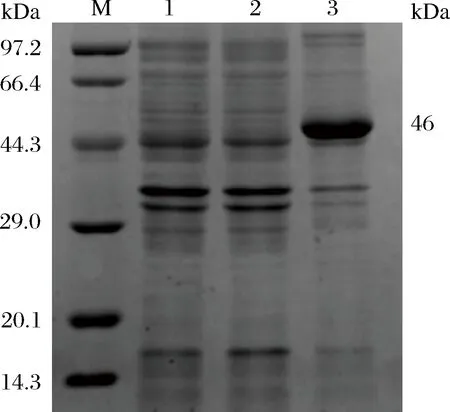
M-Marker;1-E.coli BL21;2-Eco/pEE(-IPTG); 3-Eco/pEE (+0.2 mmol/L IPTG)图2 重组蛋白的检测Fig.2 Detection of recombinant protein
2.3 H2O2浓度对大肠杆菌生长的影响
H2O2对大肠杆菌生长的影响如图3所示。在对照组中,细菌数量在前6 h内迅速增加,然后生长趋于稳定。与对照组相比,用浓度为1、2和4 mmol/L的H2O2处理大肠杆菌在培养期间显示出细胞数量的有所下降。这表明H2O2的浓度在低于最低抑菌浓度下,不能完全抑制大肠杆菌的生长,当H2O2浓度为16 mmol/L时,细菌数量明显减少且不再增加,表明剂量为16 mmol/L的H2O2能够完全抑制大肠杆菌的生长。实验表明,外源添加浓度为4 mmol/L的 H2O2产生的氧化应激效应会使大肠杆菌产生显著的生长抑制,但是不会引起细胞的死亡,因此采用4 mmol/L的H2O2作为氧化应激条件。
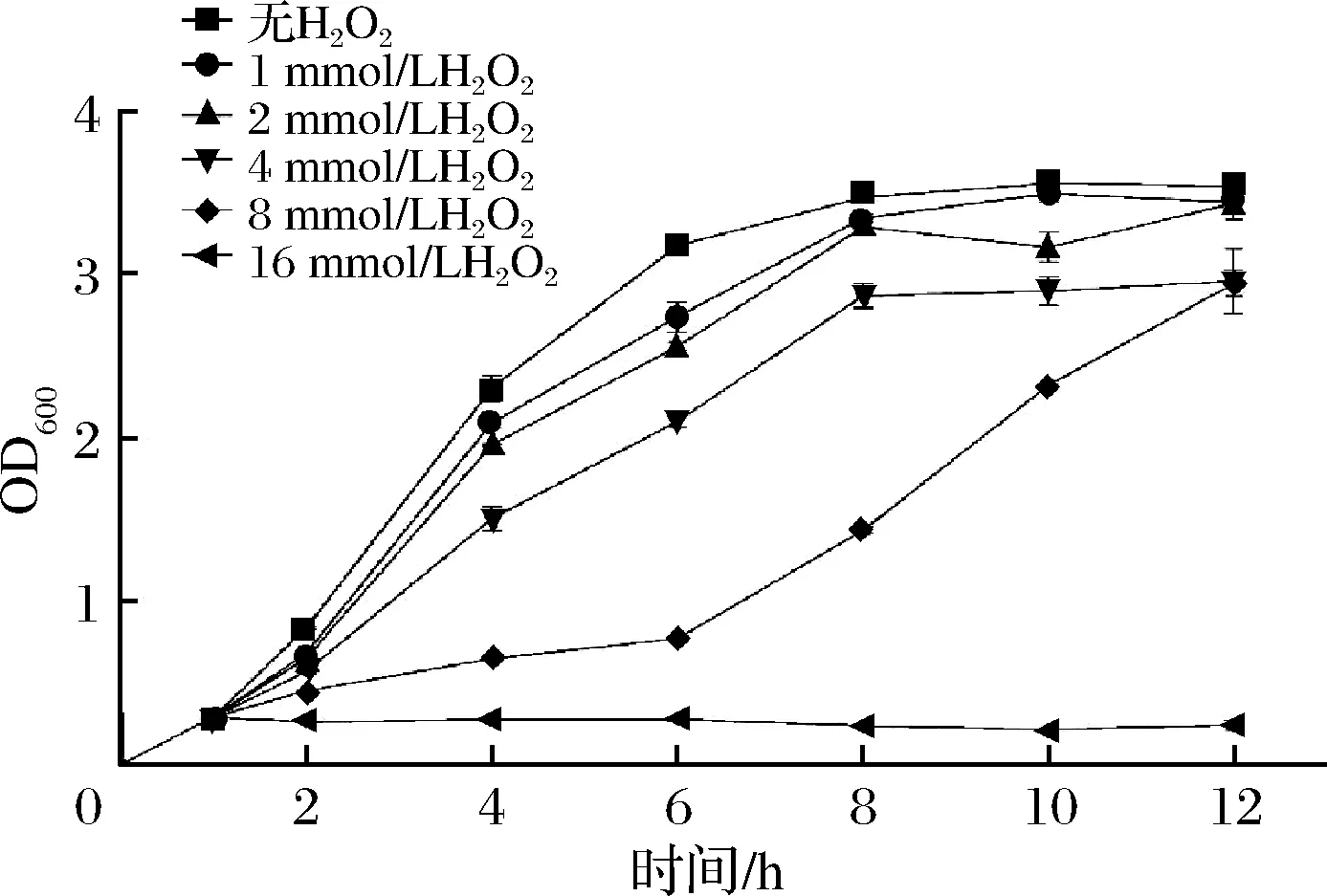
图3 用H2O2处理大肠杆菌BL21(DE3)的生长曲线Fig.3 Treatment of growth curve of E.coli BL21(DE3) with H2O2
2.4 efeB基因敲除及过表达对大肠杆菌生长的影响
为了分析efeB的缺失及过表达是否能够影响菌株的生长,测定了出发菌株与EcoΔefeB及过表达菌株Eco/pEE在正常培养及外源添加4 mmol/L H2O2刺激下的生长曲线,结果如图4所示。
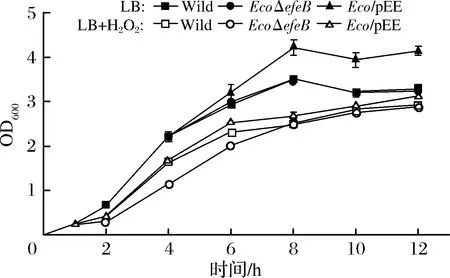
图4 不同菌株的生长曲线Fig.4 Growth curves of different strains
由图4可知,相比于出发菌株,在正常状态及H2O2刺激培养条件下,过表达efeB均促进了菌株的生长;在正常状态下,efeB的缺失未对菌体生长产生显著影响,但在H2O2刺激下,敲除菌株的生长在2~8 h明显受到抑制。以上结果表明在氧化应激状态下,EfeB蛋白在维持胞内氧化还原平衡,促进菌体生长方面发挥重要作用。
2.5 细胞膜脂质过氧化水平的测定
MDA是细胞膜受到胞内ROS攻击后的主要产物,MDA含量的变化可以反映出大肠杆菌胞内的受损程度[23]。由图5可以看出,在正常条件下,EcoΔefeB菌株与出发菌株相比,胞内MDA含量有所升高,这说明efeB的缺失加剧了胞内氧化损伤,而Eco/pEE菌株与出发菌株相比,胞内MDA含量大幅度下降,说明Eco/pEE菌株有着更强抗氧化能力,即efeB基因的表达有利于减轻胞内因氧化应激带来的损伤。另外通过外源添加H2O2发现所有菌株胞内MDA含量均有所升高,即加剧了胞内的氧化损伤水平,整体含量变化趋势与文献报道一致[24],该结果表明了efeB基因在减少胞内因ROS引起的氧化损伤方面具有重要作用。
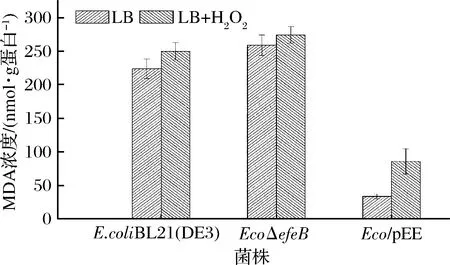
图5 不同菌株胞内脂质过氧化水平测定Fig.5 Determination of intracellular lipid peroxidation levels in different strains
2.6 胞内活性氧检测
DCFH-DA没有荧光,进入细胞后被酯酶水解为DCFH。在ROS存在时DCFH被氧化为不能透过细胞膜的强绿色荧光物质DCF,其荧光在激发波长502 nm,发射波长530 nm附近有最大波峰,强度与细胞内ROS水平成正比。用激光共聚焦显微镜定性检测胞内ROS,如图6-b所示,菌体呈现绿色荧光,表明染色成功,在此基础上用流式细胞仪进行胞内ROS定量检测,最终以荧光值来比较不同菌体之间的胞内ROS含量,结果如图7所示。
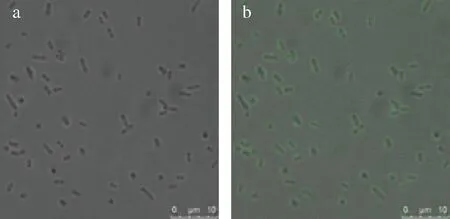
a-激光共聚焦显微镜透射光通道成像;b-激光共聚焦显微镜透射光与荧光重叠成像图6 大肠杆菌激光共聚焦显微镜染色成像Fig.6 Staining image of E.coli under laser confocal microscopy
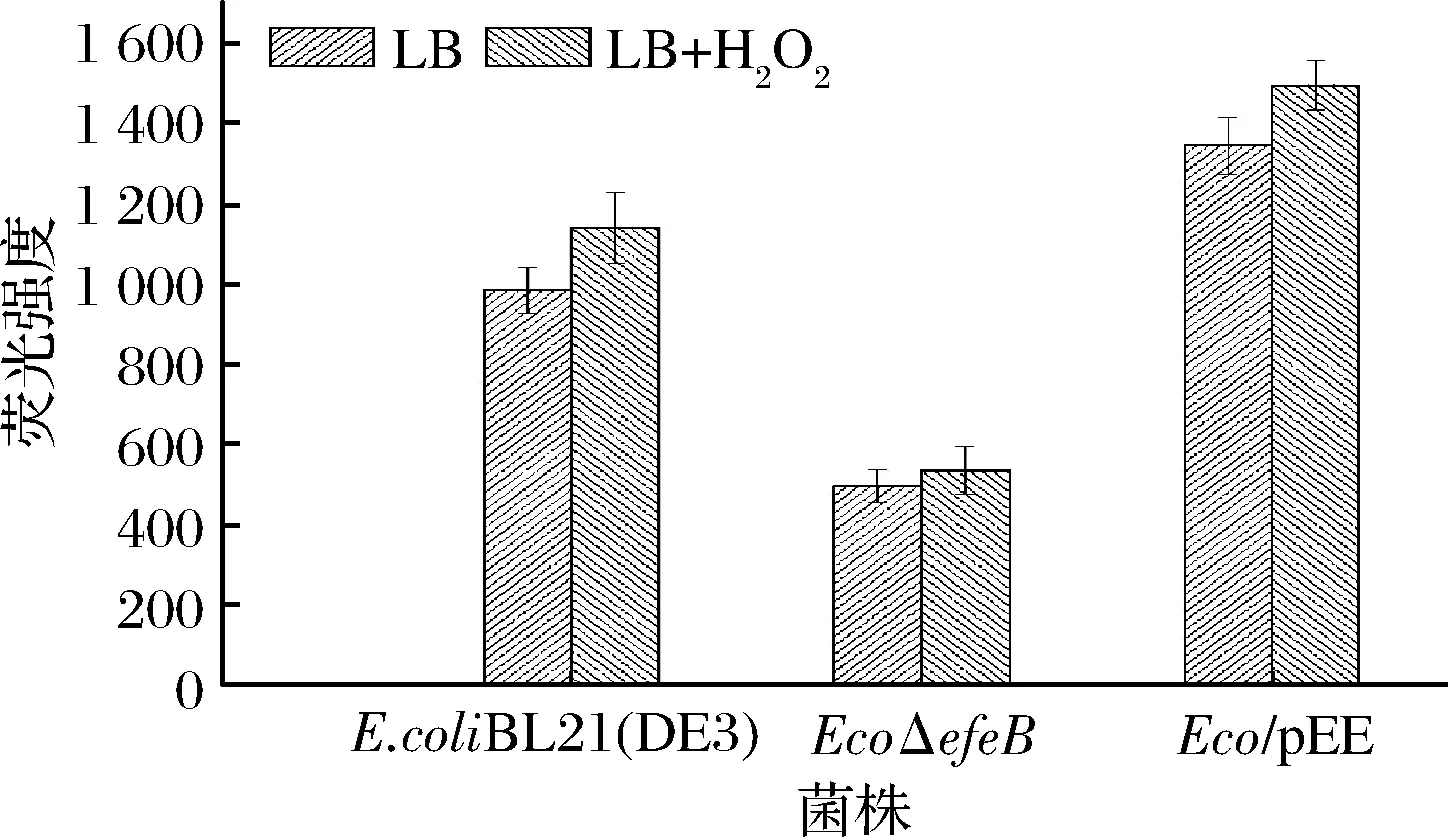
图7 不同菌株胞内ROS测定Fig.7 Determination of intracellular ROS in different strains
由图7可知,在LB培养条件下,EcoΔefeB菌株的胞内ROS含量降低,但该菌的MDA含量却是最高(图5),同时Eco/pEE菌株的胞内ROS含量升高,但对应的MDA含量却是最低(图5),结合3株菌在不同培养条件下的生长状况分析(图4),导致EcoΔefeB菌株胞内ROS含量低于出发菌株的原因可能是EcoΔefeB菌株生长缓慢,菌体呼吸活力低,呼吸链传递速率慢,同时也减少了电子泄露的可能性,从而导致EcoΔefeB菌株ROS低于出发菌株,而Eco/pEE菌体由于活力强于出发菌株,以至于Eco/pEE菌株呼吸活力高,呼吸链的电子传递速率快,但同时增加了电子泄露的可能性,导致ROS高于出发菌株[25],但由于EfeB的抗氧化应激作用,细胞膜的氧化损伤却明显低于出发菌株,表现在MDA的水平仅为出发菌株的15%(图5)。在外源添加H2O2条件下,3株菌的ROS含量进一步提升,各菌株胞内ROS含量趋势与LB正常培养条件下基本一致。
2.7 基因转录水平检测
为了探究efeB过表达对于efeB相关基因及抗氧化体系中关键基因的调控影响,对关键酶基因efeU、efeO、sodA、katE、oxyR、recA的转录水平进行了实时荧光定量 PCR分析,其基因转录水平的变化结果如图8所示。

图8 Eco/pEE菌株抗氧化途径中各基因的相对转录水平Fig.8 Relative transcript levels of each gene in the antioxidative pathway of Eco/pEE strain
在大肠杆菌中,EfeUOB系统是三联铁转运蛋白,在革兰氏阴性细菌中低pH条件下发挥铁转入及改变铁离子价态的功能[13]。从图8可以看出,efeB过表达菌株在LB 培养条件下,efeU和efeO发生了不同程度的上调,这2个基因涉及铁离子的转运,因此efeB的过表达促进了铁的转运。3个氧化应激相关基因sodA、katE和oxyR的转录水平也发生了不同程度的变化,sodA是大肠杆菌中表达超氧化物歧化酶的关键结构基因,其可以在超氧化物胁迫下保护细胞[26]。oxyR在大肠杆菌中受H2O2胁迫诱导,从而增强抗氧化防御,涉及过氧化氢酶和过氧化物酶的调控表达[27]。sodA和oxyR分别上调了2.48倍和1.95倍,这表明efeB过表达菌株为了降低ROS水平启动了其他抗氧化系统。负责过氧化氢酶的基因katE的转录水平下调了87.62%,在添加H2O2刺激后更是下调了94.81%,表明efeB基因的过表达,在维持胞内ROS的氧化还原平衡方面,尤其是H2O2的消除方面发挥了重要作用,相关机制有待进一步研究。除此之外,DNA损伤反应调节基因recA也受到了影响,recA的表达有所上调,细菌重组酶recA在DNA损伤反应中对调节DNA修复至关重要[28]。通过外源添加H2O2进一步验证,与对照组基因转录水平趋势一致。
3 结论
本研究旨在探究大肠杆菌EfeB在细胞氧化应激下的作用机制,通过测定不同菌株在正常状态及H2O2刺激条件下胞内ROS、MDA含量以及相关基因的转录情况来评估细胞内氧化应激状态的变化。结果表明大肠杆菌EfeB蛋白在消除胞内ROS及减少氧化损伤方面发挥重要作用,其中过表达efeB能够有效地调节细胞在氧化胁迫下胞内氧化还原的平衡,促进细胞生长。因此,它可能是一种安全的抗氧化剂的潜在候选者,本研究结果对其抗氧化功能的开发和利用提供了一定的理论依据。
猜你喜欢
生物学杂志(2022年5期)2022-10-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
中国调味品(2022年9期)2022-08-30
成都医学院学报(2022年4期)2022-08-19
当代水产(2022年1期)2022-04-26
中国畜禽种业(2021年11期)2021-12-09
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
中国医学影像技术(2019年6期)2019-06-24
食品界(2019年2期)2019-03-10
