
一株内毒素缺陷型大肠杆菌的构建与鉴定
2020-08-31 07:01吴骏晨乔绪稳张元鹏郑其升牛家强侯继波
中国预防兽医学报 2020年7期
吴骏晨,李 丁,乔绪稳,张元鹏,陈 瑾,郑其升*,牛家强,侯继波
(1. 西藏农牧学院 动物科学学院,西藏 林芝 860000;2. 国家兽用生物制品工程技术研究中心,江苏 南京 210014)
大肠杆菌表达系统是目前基因工程中最常使用的重组蛋白表达系统之一,因其生长速度快、易于遗传操作和高水平的重组蛋白合成率而成为多种重组蛋白表达的理想宿主之一。但复杂的LPS 纯化过程是利用该表达系统工业化生产目的蛋白,特别是兽用疫苗蛋白抗原的核心瓶颈之一[1]。
内毒素又名脂多糖(Lipopolysaccharide,LPS),一般由类脂A、核心寡糖和O-抗原构成。其中类脂A 与大肠杆菌毒力相关。通过LPS 类脂A 的介导作用,致病菌可以被宿主表面的Toll-like 受体4(Toll-like receptor 4,TLR4)识别,引起细胞反应并产生多种细胞因子,从而引起机体发热、寒战等症状。虽然,细菌LPS 一直以来都被认为是一种潜在的佐剂,但其毒性作用限制了其应用于人体[2]。Ribi等人研制了无毒力的单磷酰脂质A(MPLA),MPLA、铝佐剂和抗原组成的疫苗是目前批准的乙型肝炎疫苗和乳头瘤疫苗的主要成分,且安全有效[3-4]。LPS和MPLA 均能够特异性激活TLR4,但MPLA 仅通过β-干扰素TIR 结构域衔接蛋白(Toll-interleukin-1 receptor domain-containing adapter inducing interferon-β,TRIF)进行信号转导,而LPS 则同时通过TRIF 和髓样分化因子88(MyD88)途径传导,而后者往往会导致较高水平的炎症因子尤其是TNF-α的表达[5]。
类脂A 的合成途径相对保守,在其转运到外膜外侧的过程中其结构会发生不同的修饰作用以适应不同的外界环境。类脂A 的结构修饰在细菌体内受到非常严谨的调控且与细菌的致病性密切相关,但却不影响细菌的增殖[2,6]。基于此,本研究利用Red重组系统对大肠杆菌DH10B-2B 进行基因编辑,敲除与LPS合成相关的kdsD、lpxL、lpxM、pagP、lpxP、eptA等基因,以降低大肠杆菌LPS 的毒力[7-8],并在该大肠杆菌中表达弗朗西斯菌的LpxE 磷酸酶,后者可去除类脂A 的1-磷酸基团,从而进一步降低大肠杆菌LPS 的毒性作用[9]。本研究经鲎试剂、小鼠感染试验检测了改造大肠杆菌LPS 的毒力,同时检测了感染小鼠的细胞因子,旨在构建既可以用作低LPS 重组蛋白表达系统,也可以用于低毒力LPS 提取的大肠杆菌,为相关免疫增强剂的研究奠定基础。
1 材料与方法
1.1 主要实验材料菌种DH10B-2B 及质粒R6Kbox(内有lox71-KanR-lox66 打靶元件)、pKD46(温敏质粒)、pCP-Cre(温敏质粒)由南京农业大学崔中利老师惠赠;lpxE 基因由南京金斯瑞公司合成后亚克隆至R6K-box 中构建重组质粒lpxE-R6k-box。
高保真酶(TransStart FastPfuFly DNA Polymerase)和6×DNA Loading Buffer 购自北京全式金生物技术有限公司;2×Taq Master Mix(Dye Plus)购自南京诺唯赞生物科技有限公司;薄型琼脂糖凝胶DNA 回收试剂盒(离心柱型)和质粒小量制备试剂盒(离心柱型)购自上海捷瑞生物工程有限公司;溶菌酶、DNA 分子量标准(5 000 bp)购自生工生物工程(上海)股份有限公司;鲎试剂购自湛江安度斯生物有限公司,IL-6及TNF-a ELISA 试剂盒购自南京奥青生物科技有限公司。BALB/c小鼠购自扬州大学比较医学中心。
1.2 引物设计与合成本实验所用引物见表1,以制备敲除大肠杆菌DH10B-2B 中kdsD 基因的线性双链DNA 打靶元件的引物为例,根据GenBank 中大肠杆菌K-12 的kdsD 基因序列(947734),在其上下游约100 bp 处寻找约50 bp 的序列,并于上游50 bp 序列的5'端加上20 bp lox71 的序列,下游50 bp 的3'端加 上20 bp lox66 的 序 列(lox71 及lox66 为 质 粒R6K-box 中打靶元件的部分基因序列),最终分别合成70 bp 的引物对kdsD-F/kdsD-R。在kdsD 基因前后约200 bp 处分别截取约25 bp 的序列kdsD-YZ-F/kdsD-YZ-R 作为鉴定kdsD 基因敲除的引物。此外,从质粒R6K-box中KanR内部截取25 bp片段的kan-YZF 用于kdsD 打靶元件插入DH10B-2B 株基因组后的鉴定。其他基因敲除引物和插入基因的打靶元件设计同上。全部引物均由金斯瑞生物科技有限公司合成。

表1 本实验用到的引物Table 1 Primers used in this study
1.3kdsD线性双链DNA 打靶元件的制备以R6K-box 质粒为模板,利用kdsD-F/kdsD-R 引物对扩增kdsD 的打靶元件kdsD-Lox。PCR 产物纯化回收后由南京金斯瑞生物科技有限公司测序。其他基因的打靶元件均以R6K-box 质粒为模板同理构建。
1.4 缺失kdsD基因菌株的筛选与鉴定采用CaCl2法将编码Red 重组系统的pKD46 质粒转化至DH10B-2B,利用含有Amp+(50 μg/mL)的LB 固体培养基平板进行筛选。将经过筛选的转化子接种于200 mL终浓度含50 μg/mL Amp+和20 mmol/L L-阿拉伯糖的LB 液体培养基中,培养至OD600nm=0.4~0.6,采用电击法转化打靶元件kdsD-Lox,电转菌液涂布于含有Kan+(30 μg/mL)的LB平板上筛选阳性转化子;转化子利用kan-YZ-F/kdsD-YZ-R 引物对进行PCR 鉴定,正确的转化子命名为DH10B-2B(ΔkdsD∷kan)。
1.5lox位点重组大肠杆菌的获得与鉴定采用CaCl2法将pCP-Cre 质粒转化至DH10B-2B(ΔkdsD∷kan)中,利用含Chl+(20 μg/mL)的LB 平板筛选。将筛选的转化子接种到4 mL 含Chl+和IPTG 的LB 液体培养管中,培养约10 h,再于43 ℃过夜培养后经LB 平板划线,37 ℃培养12 h;挑取单菌落依次转接于含Kan+(30 μg/mL)、Amp+(50 μg/mL)、Chl+(20 μg/mL)和无抗生素的LB 平板上37 ℃过夜培养。挑取3 种抗性平板都不生长而只在无抗LB 平板上生长的转化子,提取其全基因组后,采用kdsD-YZ-F/kdsD-YZ-R 引物对进行PCR 鉴定。正确的转化子命名为DH10B-2B(ΔkdsD)。
1.6 其他与LPS 合成相关基因的敲除基因lpxL、lpxM、pagP、lpxP、eptA 的敲除方法同上述kdsD 基因的敲除方法,所需引物见表1。全部基因敲除后的大肠杆菌经琼脂糖凝胶电泳检测。敲除所有6 个基因的菌株命名为DH10B-2B-Δ6。
1.7lpxE基因的重组以质粒lpxE-R6k-box 为模板,lpxE-F/lpxE-R 为引物对制备打靶元件,重组方法同1.4。得到的转化子(lpxE-Lox 重组至DH10B-2B-Δ6 的OmpT 基因处)以OmpT-YZ-F/OmpT-YZ-R引物对做PCR 鉴定,正确的转化子命名为DH10B-2B-Δ6-lpxE。
1.8 改造后的大肠杆菌LPS 含量的测定将DH10B 接 种 于200 mL LB 培 养 基 中,DH10B-2B-Δ 6-lpxE 接种于200 mL Kan+抗性LB 培养基中过夜培养后经菌落计数。两种菌液分别经PBS 洗涤3 次,调整菌落数量至1.6×1010cfu/mL,将其10 被倍比稀释(108cfu/mL~105cfu/mL)后分别经20 mg/mL 溶菌酶处理,再经超声波破碎,离心后收集上清。按照鲎试剂说明书检测两种菌粗提物的LPS。
1.9 改造后的大肠杆菌对小鼠的毒力试验将45只BALB/c小鼠分为9组,5只/组,其中4组小鼠分别注射浓度为1.6×1010cfu/mL~1.6×107cfu/mL 的DH10B超声裂解物300 μL,另外4 组分别注射浓度为1.6×1010cfu/mL~1.6×107cfu/mL 的DH10B-2B-Δ6-lpxE 超声裂解物300 μL,最后一组注射等量无菌PBS 作为对照组。观察各组小鼠24 h 内的临床症状。
1.10 小鼠脾脏细胞因子的检测上述细菌裂解物注射小鼠24 h 后迫杀采集其脾脏样品于2 mL EP 管中,加入生理盐水,置冰上研磨均匀,离心15 min后收获上清,采用ELISA 试剂盒分别检测IL-6、TNF-α的含量。
2 结 果
2.1 线性双链DNA 打靶元件的制备结果以质粒R6k-Box 为模板,kdsD-F/kdsD-R 为引物,对制备的打靶元件进行PCR 鉴定,结果显示得到约为1 000 bp 的目的条带,与预期一致;其余打靶元件照此制备(图1)。测序结果显示打靶元件均与模板一致。表明各打靶元件正确制备。

图1 打靶元件的制备Fig.1 Preparation of targeting elements
2.2 缺失kdsD基因菌株的筛选与鉴定结果将打靶元件kdsD-Lox 电转化至已转化了pKD46 质粒的DH10B-2B 菌中,涂布于Kan+平板筛选出重组菌,利用kan-YZ-F/kdsD-YZ-R 引物对其经PCR 鉴定,结果显示扩增出约为1 200 bp 的目的条带,与预期一致(图2),经测序鉴定序列正确。表明打靶元件重组至DH10B-2B 的基因组中,得到重组菌DH10B-2B(ΔkdsD∷kan)。

图2 重组菌株DH10B-2B(ΔkdsD∷kan)的PCR 鉴定结果Fig.2 The PCR identification of the recombinant strains DH10B-2B(ΔΔkdsD∷kan)
2.3lox位点重组菌株的获得与鉴定结果将pCP-Cre 质粒转化至DH10B-2B(ΔkdsD∷kan) 中,在IPTG 诱导下,pCP-Cre 质粒可表达Cre 重组酶诱导lox71 和lox66 位点重组并失去其中的KanR,再通过43 ℃培养使温敏质粒pKD46 和pCP-Cre 丢失,获得不含任何抗性基因的lox 位点重组菌,经PCR 鉴定仅能在无抗LB 上生长的菌落。结果显示,重组菌株在约500 bp 出现目的条带,而DH10B-2B 经PCR 扩增得到约1500 bp 左右的条带(图3),表明kdsD 基因已经敲除。得到的重组菌株命名为DH10B-2B(ΔkdsD)。

图3 重组菌株DH10B-2B(ΔkdsD)的PCR 鉴定结果Fig.3 PCR identification of the recombinant strains DH10B-2B(ΔkdsD)
2.4 其它LPS 合成相关基因的敲除结果其它基因的敲除同kdsD 基因的敲除方法,各基因全部敲除后的菌株以各自的引物进行PCR 鉴定。结果显示,PCR 扩增得到的条带均位于500 bp 左右,而野生型大肠杆菌DH10B-2B 的PCR 扩增则得到1 000 bp~2 000 bp的目的条带(图4)。表明大肠杆菌DH10B-2B中6 个LPS 合成相关基因均已经被正确敲除,最终得到重组菌DH10B-2B-Δ6。
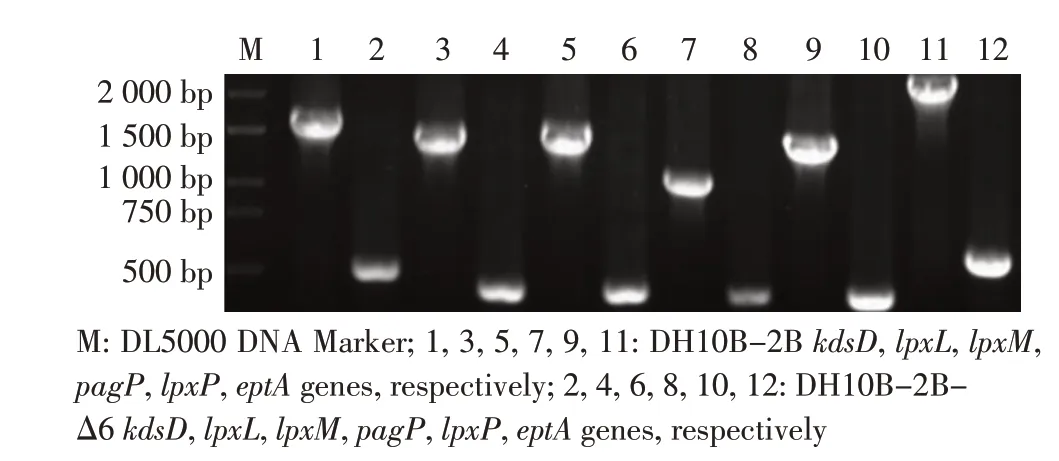
图4 重组菌株DH10B-2B-Δ6 的PCR 鉴定结果Fig.4 PCR identification of the recombinant strains DH10B-2B-Δ6
2.5lpxE基因重组的鉴定结果以1.4 的方法将lpxE-Lox 重组至DH10B-2B-Δ6 的OmpT 基因处,采用引物OmpT-YZ-F/OmpT-YZ-R 对获得的重组菌进行PCR 鉴定。结果显示,重组菌DH10B-2B-Δ 6-lpxE 扩增得到的片段约为2 200 bp,而野生型DH10B-2B 经PCR 扩增得到的条带约1 200 bp(图5)。表明lpxE 基因已经正确插入到重组菌株DH10B-2B-Δ6 的OmpT 基因处。

图5 重组菌株DH10B-2B-Δ6-lpxE 的PCR 鉴定结果Fig.5 PCR identification of the recombinant strains DH10B-2B-Δ6-lpxE
2.6 改造后E. coliLPS 的检测结果采用鲎试剂检测改造后的DH10B-2B-Δ6-lpxE 粗提物LPS。由于检测菌的浓度较大,无法测算LPS 具体含量,故仅展示LPS 的阴阳性结果。结果显示,106cfu/mL 野生型DH10B-2B 裂解液LPS 检测结果为阳性, 而106cfu/mL DH10B-2B-Δ6-lpxE 裂解液LPS 结果为阴性,108cfu/mL DH10B-2B-Δ6-lpxE 裂解液检测结果为阳性(表2)。表明,相较于野生型DH10B-2B,DH10B-2B-Δ6-lpxE 产生LPS 的能力明显减弱。

表2 鲎试剂检测结果Table 2 The results of tachypleus amebocyte lysate test
2.7 改造后的大肠杆菌对小鼠的毒力试验结果小鼠注射1.6×1010~1.6×107cfu/mL 改造前后的大肠杆菌裂解液均未出现死亡,但注射1.6×1010cfu/mL 野生型大肠杆菌裂解液的小鼠(5/5)在24 h 内相继出现腹泻、被毛蓬乱、精神沉郁、运动能力和应激反应减弱,眼角有脓性分泌物;而注射相同剂量DH10B-2B-Δ6-lpxE 的小鼠(2/5)在24 h 内仅出现软便、精神略差、被毛松散的症状。注射1.6×109cfu/mL 野生型大肠杆菌裂解液的小鼠(4/5)在24 h 内相继出现腹泻、被毛蓬乱、精神萎靡的症状,而注射相同剂量DH10B-2B-Δ6-lpxE 裂解液的小鼠在24 h 内未出现明显的症状。表明改造后的大肠杆菌产生的LPS 对小鼠的毒力明显降低。
2.8 小鼠脾脏细胞因子的检测结果利用ELISA方法检测各组小鼠脾脏中的IL-6 与TNF-α炎性细胞因子水平。结果显示,DH10B-2B-Δ6-lpxE 裂解物注射小鼠后其脾脏产生的IL-6 和TNF-α的含量均随着注射剂量的增加而增加,虽然均高于注射PBS 的对照组小鼠,但相较于注射同等剂量野生型DH10B-2B 裂解液的小鼠,其分泌细胞因子的能力明显降低(p<0.05;p<0.01)(图6)。结果表明相较于野生型DH10B-2B,DH10B-2B-Δ6-lpxE 的LPS 引起的炎症反应更加轻微,其毒力明显减弱,具有用于疫苗佐剂的潜力。

图6 各组大肠杆菌裂解物感染小鼠后的细胞因子检测结果Fig.6 Cytokine detection results of mice infected with E.coli lysates in each group
3 讨 论
大肠杆菌是一种常见的革兰氏阴性细菌,因其生长快、培养廉价、基因操作简单快捷、易于规模化发酵的特点被广泛应用于重组蛋白的生产[10]。然而大肠杆菌外膜中大量存在的LPS 作为一种强力免疫刺激分子,进入动物体内能诱导机体产生多种生物活性因子,从而发生感染性休克[11]。因此通过大肠杆菌生产的重组蛋白必须将LPS 去除后才能投入使用,这大大增加了开发重组蛋白产品的工序和成本。
为构建一株低毒力的大肠杆菌用作重组蛋白表达体系或用于提取低毒力的LPS 。本研究通过Red和Cre-Lox 敲除系统相结合的方法,敲除大肠杆菌DH10B-2B 株中与LPS 合成相关的基因(kdsD、lpxL、lpxM、pagP、lpxP、eptA),并在其OmpT 基因处插入弗朗西斯菌的磷酸酶基因lpxE,得到LPS 缺陷型大肠杆菌DH10B-2B-Δ6-lpxE。kdsD 基因调控合成LPS 中3-去氧-D-甘露-2-辛酮糖酸(Kdo)的关键酶的合成;LpxL、LpxM 分别在LPS 远端的氨基葡萄糖中添加一个次级月桂酰残基和一个豆蔻醇残基;PagP 将棕榈酸酯从甘油磷脂转移到类脂A 的β-2 位置,形成七酰化结构;LpxP 与LpxL 具有很高的相似度,在低温条件下以棕榈油酸取代月桂酸转移到脂质A2'位置;EptA 则可在细菌内膜外表面将磷酸乙醇胺转移至类脂A[12-13]。研究表明,敲除这些基因后可降低LPS 对机体的免疫刺激作用,降低细菌感染带来的风险[14-16]。通过将弗朗西斯菌的lpxE 基因整合到大肠杆菌的基因组中使其表达LpxE酶,可以去除LPS 中的类脂A1-磷酸基团,使大肠杆菌表达出仅含一个磷酸基团的类脂A,即单磷酸脂质A(MPLA)[17]。MPLA 是一种仅含一个磷酸基的脂质A 衍生物,在保留了许多免疫调节特性的同时其毒力比野生型脂质A 小得多[18-19]。
鲎试剂检测结果表明DH10B-2B-Δ6-lpxE 裂解液中LPS 的含量要远低于野生型细菌。与野生型细菌相比,感染相同浓度DH10B-2B-Δ6-lpxE 裂解液的小鼠症状更轻微,恢复的更快。并且ELISA 结果显示改造后的细菌刺激小鼠产生IL-6 及TNF-a 的水平明显减弱。而通常游离的LPS 进入机体后可以激活TLR4,刺激TRIF 和MyD88 信号通路,产生较高水平的炎症因子尤其是TNF-α及IL-6 等[11]。以上结果 均 表 明DH10B-2B-Δ6-lpxE 的LPS 毒 力 相 较 于DH10B-2B 明显减弱。
本研究通过敲除与大肠杆菌LPS 合成相关的酶基因,并插入lpxE 基因去除了LPS 类脂A 中的A1-磷酸基团,构建了LPS 缺陷型重组菌DH10B-2BΔ6-lpxE,该菌产生的LPS 毒力显著降低,为其以后作为低毒力大肠杆菌表达体系及用于提取减毒LPS作为疫苗免疫增强剂研究奠定了基础。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中国特种设备安全(2022年1期)2022-04-26
检验医学(2021年11期)2021-11-29
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
农药科学与管理(2019年9期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
中国抗生素杂志(2019年6期)2019-07-06
中国核电(2017年2期)2017-08-11
