
miR-214 通过激活肝星状细胞促进肝纤维化进程
2020-08-18 10:06:00公绪华钱黎俊
上海交通大学学报(医学版) 2020年6期
公绪华,朱 樑,陈 超,钱黎俊
1. 上海交通大学医学院附属仁济医院放射科,上海 200127;2. 上海长征医院消化内科,上海 200003;3. 中国人民解放军总医院第四医学中心消化内科,北京 100048
肝纤维化是细胞外基质(extracellular matrix,ECM)发生弥漫性过度沉积的一种病理过程,在大多数慢性肝病中尤为常见[1]。肝星状细胞(hepatic stellate cells,HSCs)是肝脏特有的一种非实质细胞,仅占肝脏细胞总数的5%~7%。研究[2]显示,HSCs 是ECM 的主要来源细胞,也是肝纤维化的主要效应细胞,且该细胞的激活是肝纤维化的关键环节。当肝脏受到损伤时,静息状态的HSCs 则被激活,从而可增加其增殖、收缩、迁移能力,同时也可增加ECM 的分泌能力[3]。活化型HSCs 可表达α-平滑肌肌动蛋白(α-smooth muscle actin,α-SMA),并分泌Ⅰ、Ⅲ、Ⅳ型胶原等ECM 成分,这些成分可沉积于门脉周围、Disse 间隙及肝小叶内,形成“肝窦毛细血管化”;同时,活化型HSCs 收缩力增强可增加肝窦血流阻力,增加门脉压力,从而使肝内结构重建,进而导致肝纤维化[4]。
miRNAs 是一类在转录后水平调节基因表达的非编码RNA,参与细胞的代谢、增殖、分化以及凋亡等生理过程[5]。目前已发现有多种miRNAs 在HSCs 活化和肝纤维化过程中发挥作用,如miR-221/222、miR-15b、miR-16等[1,6],但miRNAs 调控HSCs 活化的分子机制及其水平与临床肝纤维化分期的相关性尚未被完全阐明。
既往相关报道[7]显示,miR-214 在甲硫氨酸和胆碱缺乏饮食诱导的肝纤维化模型中表达升高。我们推测miR-214 可能在HSCs 的活化及肝纤维化进程中发挥重要作用。基于此,本研究对miR-214 参与的HSCs 活化过程中的生物学功能及其可能的作用机制进行探讨,以期对肝纤维化的基因治疗提供新的思路。
1 对象与方法
1.1 实验对象
1.1.1 实验动物 选取雄性Wistar 大鼠40 只[由海军军医大学动物科学部提供,动物生产许可证号:SCXK(沪)2012-0002],体质量为180 ~200 g。大鼠饲养于海军军医大学动物中心清洁级动物房的标准笼中,动物使用许可证号:SYXK(沪)2013-0050。大鼠饲以标准饲料,自由摄食、饮水。饲养条件如下:光周期为12 h/12 h,温度为25 ℃左右,湿度为50%~60%,环境噪声≤60 dB。动物饲养条件符合实验动物管理条例,所有动物相关操作均遵循国家及海军军医大学实验动物科学部有关动物伦理规定及条例。
1.1.2 细胞株、主要试剂及仪器 大鼠HSC 细胞株HSC-T6 及293T 细胞均由上海长征医院消化内科实验室保存。TRIzol-A+总RNA 提取试剂购自天根生化科技(北京)有限公司,Transwell 小室购自美国EMD Millipore 公司,小鼠α-SMA 单克隆抗体均购自德国Sigma-Aldrich 公司,兔抗1 型胶原蛋白(collagen type 1, COL1)多克隆抗体、α-tubulin 抗体、GAPDH 抗体、兔抗缺氧诱导因子-1α 抑制剂(hypoxia-inducible factor-1α inhibitor,HIF1AN)多克隆抗体均购自美国Abcam 公司,反转录和实时荧光定量PCR(quantitative real-time PCR,qPCR)试剂盒、双荧光素酶报告基因实验试剂盒均购自美国Promega 公司,慢病毒载体表达系统购自上海生博生物医药科技有限公司。miR-214 模拟物(miR-214 mimics)、含无义序列模拟物(NC mimics)、miR-214 抑制剂(miR-214 inhibitor)及双荧光素酶报告基因实验所需质粒[HIF1AN-WT 质粒(野生型)、HIF1AN-MuT 质粒(突变型)]均由武汉谷歌生物科技有限公司合成。
LightCycler®96 实 时 荧 光 定 量PCR 仪 购 自 瑞 士Roche 公司,FACSCalibur 流式细胞仪购自美国Bection Dickinson 公司。
1.2 实验方法
1.2.1 大鼠分组及肝纤维化模型构建 本研究采用经典的CCl4诱导大鼠肝纤维化的模型进行实验[8],具体步骤如下:将40 只大鼠随机分为对照组(即con 组)和造模组,每组20 只。向造模组大鼠腹腔注射体积分数20%的CCl4油剂溶液,每周2 次,每次剂量2.5 mL/kg,连续4 周;同时,向con 组大鼠注射等体积的无菌油剂。分别于不同时间(第1、2、3、4 周末)处理2 组大鼠(每组5 只),留取肝脏组织标本。通过苏木精-伊红染色(H-E 染色),于光学显微镜下观察大鼠肝纤维化进程。
1.2.2 大鼠原代HSCs 的分离与培养 经前期实验[9]证实,大鼠原代HSCs 的分离纯度及活性均可达到95%以上。
经2 个消化步骤从健康雄性Wistar 大鼠肝脏分离获得原代HSCs。采用经典的密度梯度离心法进行分离。而后,采用含10%胎牛血清(fetal bovine serum,FBS)、1%的青霉素/链霉素、2 mmol/L 谷氨酰胺的高糖DMEM 培养基培养已纯化的原代HSCs,分别于第2、7、14 日收集HSCs 用于后续研究。将体外培养2 d 的原代HSCs 作为静息状态HSCs,培养7 d 作为部分活化状态HSCs,培养14 d 作为完全活化状态HSCs。
1.2.3 慢病毒感染HSC-T6 细胞及分组 慢病毒的包装和生产依据操作说明进行。培养HSC-T6 细胞至对数生长期,用0.25%胰蛋白酶进行消化、传代,按1×106/孔接种于6 孔板。于超净工作台内,将携带有绿色荧光蛋白(green fluorescence protein,GFP)的慢病毒颗粒按感染复数(multiplicity of infection,MOI)=30 加入相应孔内,12 h 后更换新鲜DMEM 培养基,继续培养3 d。依据慢病毒携带序列的不同,将感染后的HSC-T6 细胞分为miR-214 过表达组(lenti-pLVT525-miR-214)、miR-214 敲除组(lenti-pLKO.1- miR-214)和阴性对照慢病毒组(lentipLVT4,即mock 组)。
1.2.4 反转录与qPCR 检测 用TRIzol-A 提取原代HSCs、大鼠肝脏组织、慢病毒转染组HSC-T6 细胞的总RNA,按照反转录试剂盒说明书合成cDNA,以cDNA 为模板进行PCR 反应。扩增条件为:95 ℃ 2 min;95 ℃ 15 s,60 ℃ 30 s,70 ℃ 1 min,共40 个循环。设置3 个复孔,记录每孔的CT值,以3 个复孔的平均值作为最终结果。本研究中miRNA 的qPCR 方法为加尾法,以U6 为内参,采用2-△△CT法进行分析。引物序列见表1。

表1 qPCR 引物序列Tab 1 Primer sequences for qPCR
1.2.5 蛋白质印迹检测 分别收集miR-214 过表达组、miR-214 敲 除 组 及mock 组 的HSC-T6 细 胞,4 800×g离心2 min 后弃去上清液,用磷酸盐缓冲液(phosphate buffered saline,PBS)洗涤2 ~3 次,并用裂解液充分裂解细胞,4 800×g 离心10 min 后弃去沉淀,获得总蛋白。经凝胶电泳、硝酸纤维素膜印迹后,用含5%脱脂奶粉的TBST 封闭2 h。而后将膜转移至含有α-tubulin 抗体、GAPDH 抗体、COL1 抗体、α-SMA 抗体及HIF1AN 抗体(工作浓度均为1:1 000)的稀释液中,于4 ℃孵育过夜。再用TBST 洗涤3 次(每次10 ~15 min)后,将膜与相应二抗常温孵育2 h,TBST 洗涤3 次。最后,用ECL 化学发光试剂显影。
1.2.6 细胞迁移检测 该实验于24 孔Transwell 组织培养板中进行。采用胰蛋白酶消化miR-214 过表达组、miR-214 敲除组及mock 组的HSC-T6 细胞,PBS 洗涤2 次,再用含1%FBS 的DMEM 培养基重悬,制备4×105个/mL 的细胞悬液。向24 孔板小室中加入细胞悬液,每孔100 μL,小室外层浸润在600 μL 含10%FBS 的DMEM 培养基中,于37 ℃、5% CO2条件下培养20 h 后用4%多聚甲醛固定,室温下用Hoechst 33342 染色5 min,并用棉签除去未迁移的细胞,于免疫荧光显微镜下随机选取4 个视野进行观察。
1.2.7 细胞凋亡检测 分别取约5×105个miR-214 过表达组、miR-214 敲除组及mock 组的HSC-T6 细胞于1.5 mL 离心管中,用预冷的PBS 清洗3 次,按凋亡试剂盒说明书制备混悬液,避光水浴5 min 后采用流式细胞仪对细胞凋亡情况进行检测。
1.2.8 双荧光素酶报告基因实验检测 TargetScan 靶基因预测数据库预测Hif1an 为miR-214 的潜在靶基因。向293T 细胞分别转染HIF1AN-WT 质粒(表达野生型HIF1AN,WT 组)和HIF1AN-MuT 质粒(表达突变型HIF1AN,MuT 组),再利用脂质体向上述293T 细胞分别转染miR-214 mimics 和NC mimics,而后向转染miR-214 mimics 的293T 细胞中再加入miR-214 抑制剂,因此共获得6 组, 即miR-214 mimics+HIF1AN-WT 组、miR-214 mimics+inhibitor +HIF1AN-WT 组、NC mimics+ HIF1ANWT 组、miR-214 mimics+ HIF1AN-MuT 组、miR-214 mimics+inhibitor + HIF1AN-MuT 组、NC mimics+ HIF1AN-MuT 组。继续培养48 h,于酶标仪双荧光素酶报告基因检查系统检测上述6 组细胞的荧光素酶活性,结果取平均值。
1.3 统计学方法
采用SPSS 17.0 软件对研究数据进行统计分析。定量资料以±s 表示,采用t 检验进行2 组间比较,采用方差分析进行2 组以上比较。P<0.05 表示差异具有统计学 意义。
2 结果
2.1 miR-214 在HSCs 活化和肝纤维化过程中的表达变化
对con 组及造模组大鼠的肝脏组织进行H-E 染色,以确定肝纤维化分期。经病理检查结果证实,随着时间推移肝纤维化程度逐渐加重(图1A)。
对纯化后的大鼠原代HSCs 培养发现,HSCs 初始为形态均一的小圆细胞;经体外培养2 d 后,多数呈贴壁生长,形态呈椭圆形;培养7 d 后,其胞质开始伸展,胞体变得细长;14 d 后其胞质完全伸展,细胞内可见平行排列的细胞骨架(图1B)。
经qPCR 检测显示,miR-214 在原代HSCs 活化过程及CCl4诱导的大鼠肝纤维化进程中表达逐渐增加,且差异具有统计学意义(均P<0.05,图1C);同时,在原代HSCs 活化过程中,HSCs 活化标志物Col1 mRNA 及α-Sma mRNA 表达亦逐渐增加(均P<0.05,图1D)。
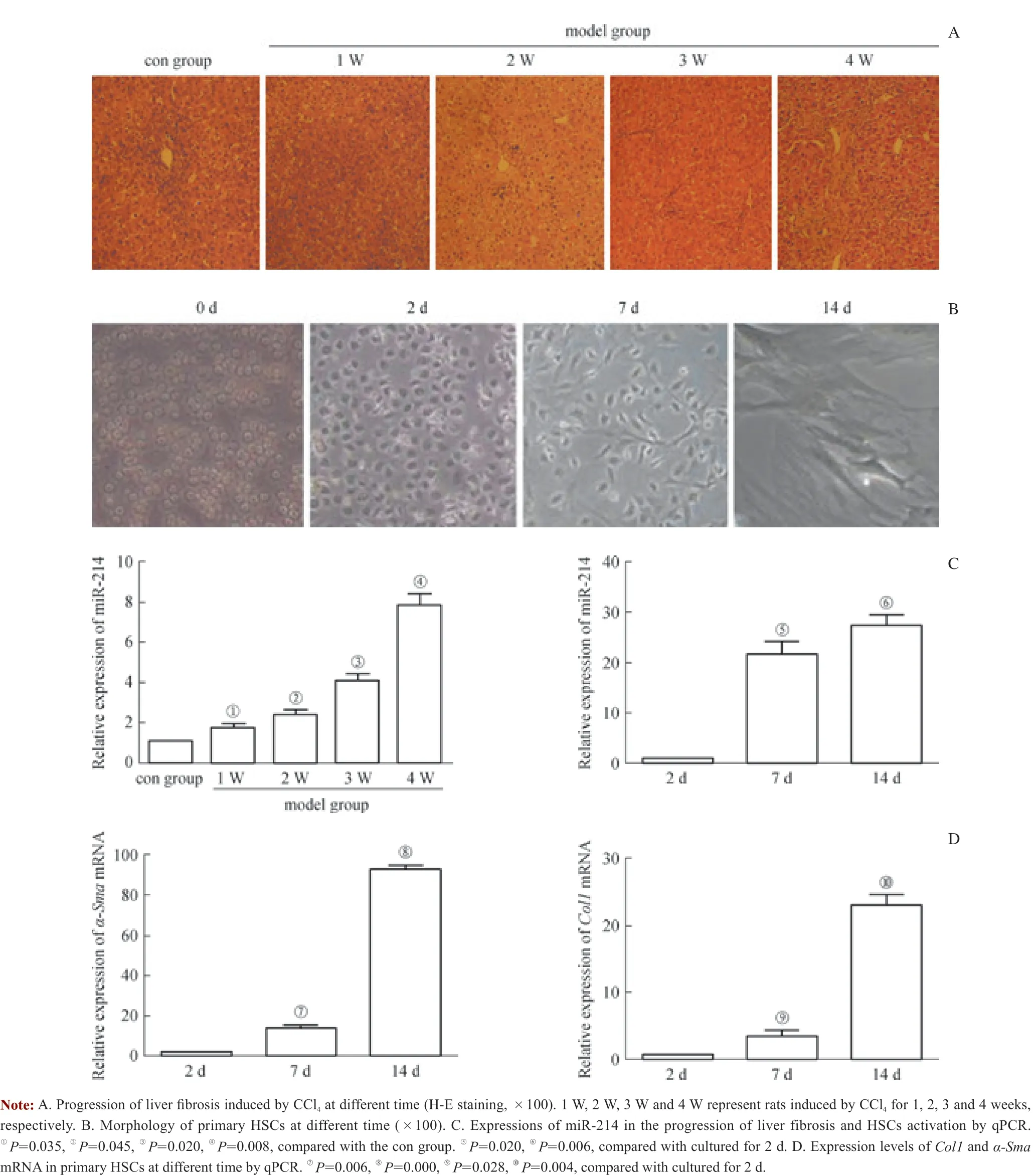
图1 miR-214 在肝纤维化进程和HSCs 活化过程中的表达变化Fig 1 Expression of miR-214 in the progression of liver fibrosis and HSCs activation
2.2 慢病毒转染组HSC-T6 细胞中α-SMA 及COL1 的表达变化
本研究中MOI=30,通过免疫荧光显微镜观察经慢病毒转染的HSC-T6 细胞中GFP 亮度并计算荧光细胞比例,结果显示慢病毒转染效率约为90%。qPCR 检测显示,与mock 组相比,miR-214 过表达组中miR-214 的表达量增加近3 倍(P=0.000),继而证实miR-214 过表达慢病毒载体构建成功(图2A)。
随 后,采 用qPCR 对α-SMA 及COL1 的 表 达 进 行检测,结果显示,与mock 组相比,miR-214 敲除组中α-Sma 及Col1 mRNA 的表达水平有所下降,而miR-214过表达组则有所升高(均P<0.05,图2B)。同样的,蛋白质印迹法检测慢病毒转染组HSC-T6 细胞中α-SMA 及COL1 蛋白的表达,显示出了相同的结果(均P<0.05,图2C)。继而提示,miR-214 可促进HSCs 的活化。

图2 慢病毒转染组HSC-T6 细胞中α-SMA 及COL1 的表达变化Fig 2 Expression of α-SMA and COL1 in HSCs transfected with lentivirus
2.3 慢病毒转染组HSC-T6 细胞的迁移及凋亡检测
Transwell 细胞迁移实验(图3A)显示,与mock 组相比,miR-214 敲除组中HSC-T6 细胞的迁移数减少约50%,而miR-214 过表达组则是其2.8 倍。
为检测miR-214 是否影响HSC-T6 细胞的凋亡,本研究使用Annexin V-FITC/PI 染色进行流式细胞术分析。结果(图3B)显示,与mock 组相比,miR-214 过表达组细胞具有较高的存活能力。综合上述结果发现,miR-214 可增强HSCs 的迁移能力、降低细胞凋亡率。
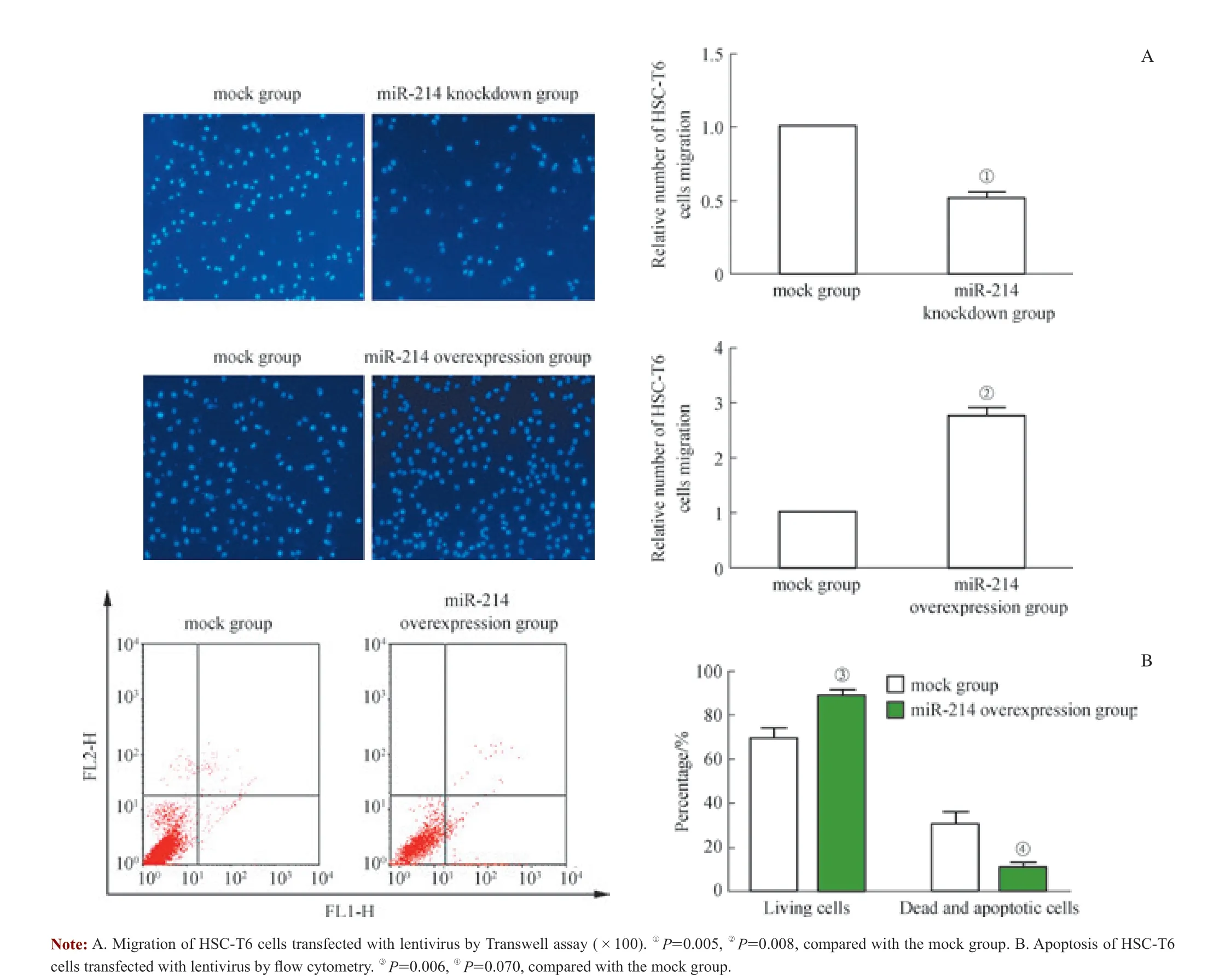
图3 慢病毒转染组HSC-T6 细胞的迁移及凋亡检测Fig 3 Detection of the migration and apoptosis of HSC-T6 cells transfected with lentivirus
2.4 miR-214 的靶基因Hif1an 在HSCs 活化和肝纤维化进程中的表达变化
通过在线检索TargetScan 靶基因预测数据库的相关信息发现,Hif1an 可能是miR-214 的潜在靶基因,同时预测出miR-214 与Hif1an 的3' UTR 的结合位点(图4A)。
双荧光素酶报告基因实验检测结果(图4 B)显示,在WT 组293T 细胞中,miR-214 mimics 能够显著抑制胞内荧光素酶活性,与NC mimics 组相比较,miR-214 mimics 组荧光素酶活性明显下降(P=0.000),而 miR-214 mimics+inhibitor 组的荧光素酶活性则不变;在MuT 组细胞中,miR-214 mimics 对胞内荧光素酶活性则无明显影响,miR-214 mimics 组与NC mimics 组的荧光素酶活性间无明显差异,继而说明RNA 转染本身对荧光素酶活性无影响。
采用qPCR 和蛋白质印迹对慢病毒转染组HSC-T6 细胞中HIF1AN 的表达进行分析,结果(图4 C、D)显示,与mock 组相比,miR-214 敲除组中HIF1AN 的mRNA 及蛋白表达量有所增加,miR-214 过表达组的HIF1AN 的mRNA 及蛋白表达量有所减少(均P<0.05)。
采用qPCR 检测原代HSCs 活化过程及CCl4诱导的大鼠肝纤维化进程中Hif1an mRNA 的表达水平,结果(图4 E、F)显示,作为miR-214 的靶基因,Hif1an 在原代HSCs 活化过程和肝纤维化进程中的表达明显下调。
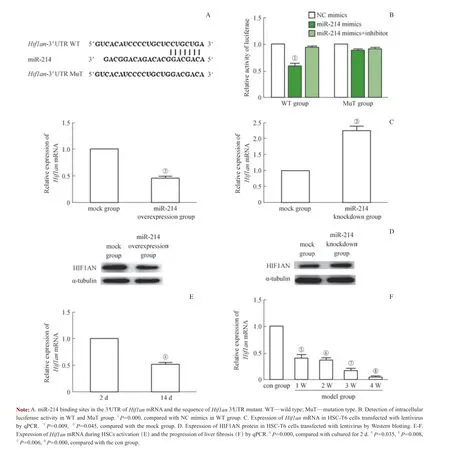
图4 miR-214 的靶基因Hif1an 在HSCs 活化和肝纤维化进程中的表达变化Fig 4 Expression of Hif1an, the target gene of miR-214, during HSCs activation and the progression of liver fibrosis
3 讨论
肝纤维化的分子机制探索及临床诊治研究是该领域在基础医学和临床医学方面所面临的重大课题。目前已经发现多种miRNAs 在HSCs 活化过程和肝纤维化进程中发挥重要作用,但具体分子机制及与肝纤维化分期的相关性尚未明确。
既往针对HSCs 活化过程中miRNAs 芯片表达谱的研究[8]提示,miR-214 在HSCs 活化过程中可表达上调。本研究发现,miR-214 在原代HSCs 活化过程及CCl4诱导的大鼠肝纤维化进程中均表达上调,这与既往文献报道的miR-214 在甲硫氨酸胆碱缺乏饮食小鼠模型[7]和二甲基亚硝胺诱导的肝纤维化大鼠模型[10]中表达升高相一致。然而,针对毒素性肝损伤的研究[7,11]显示,miR-214 的表达有所下调。这些研究表明,miR-214 水平与肝纤维化的相关性在不同的肝纤维化模型中存在一定的差异;继而提示在未来有关miRNAs 介导的肝纤维化调控研究中,需仔细考虑用于诱导肝纤维化的方法。
miRNAs 的生物学功能是通过调控其靶基因来实现的[12-13]。miR-214 在肝纤维化中的作用已有报道,但目前尚未有Hif1an 作为其靶基因的相关研究。在本研究中,我们将Hif1an 确定为miR-214 的一个新靶点。该基因是缺氧诱导因子1α(hypoxia-inducible factor-1α,HIF-1α)的抑制剂,而HIF-1α 是缺氧应激反应中的重要基因。HIF1AN 可通过羟基化HIF-1α 转录活性区域,抑制其调控下游基因[14]。近年来的研究[15-18]提示,HIF-1 在缺氧条件下可通过PTEN/p65 信号通路、NF-κΒ 途径及TGF-β/smad3 信号通路促进HSCs 活化以及肝纤维化的发生与发展。HIF-1 可参与调控诸多肝纤维化相关基因的转录,如胰岛素样生长因子(insulin-like growth factor,IGF)、内皮素-1(endothelin-1,ET-1)、血管内皮生长因子(vascular endothelial growth factor,VEGF)、 瘦 素、 基 质 金 属 蛋白 酶-2(matrix metalloproteinase-2,MMP-2) 等, 以 促进HSCs 活化以及肝纤维化进程[19-20]。既往研究[21]提示,HIF-1 在HSCs 活化和肝纤维化进程中的表达明显升高,与肝纤维化程度呈正相关,而HIF-1 的沉默则能显著抑制HSCs 的活化及肝纤维化进展,因此HIF-1 或也成为肝纤维化治疗中的潜在靶点。本研究尚存在不足之处,比如在Hif1an 作为miR-214 靶基因的论证中未涉及相关信号通路的研究等。但本课题组的后续研究发现,miR-214 基因的启动子中含有smad3 的结合元件,而smad3 作为转化生长因子β(transforming growth factor-β,TGF-β)下游的转录因子可诱导miR-214 在HSCs 中的表达,抑制miR-214 的表达则能够拮抗TGF-β/smad3 信号通路诱导的HSC 活化(待发表)。TGF-β 是肝纤维化病变过程中关键的细胞因子之一。其中TGF-β/smad3 信号通路是TGF-β 信号转导调节HSCs 活化促进ECM 生成的主要途径[22]。由此我们推测,miR-214/HIF1AN 可能参与了TGF-β/smad 通路刺激的HSCs 活化过程,同时miR-214 有望成为肝纤维化治疗中的一种特异miRNA,其相关作用机制有待更深入的研究。
参·考·文·献
[1] Ogawa T, Enomoto M, Fujii H, et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis[J]. Gut, 2012, 61(11): 1600-1609.
[2] Trautwein C, Friedman SL, Schuppan D, et al. Hepatic fibrosis: concept to treatment[J]. J Hepatol, 2015, 62(1): S15-S24.
[3] Török NJ. Recent advances in the pathogenesis and diagnosis of liver fibrosis[J]. J Gastroenterol, 2008, 43(5): 315-321.
[4] Soon RK Jr, Yee HF Jr. Stellate cell contraction: role, regulation, and potential therapeutic target[J]. Clin Liver Dis, 2008, 12(4): 791-803.
[5] Yuan K, Orcholski M, Tian XF, et al. MicroRNAs: promising therapeutic targets for the treatment of pulmonary arterial hypertension[J]. Expert Opin Ther Targets, 2013, 17(5): 557-564.
[6] Guo CJ, Pan Q, Li DG, et al. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: an essential role for apoptosis[J]. J Hepatol, 2009, 50(4): 766-778.
[7] Iizuka M, Ogawa T, Enomoto M, et al. Induction of microRNA-214-5p in human and rodent liver fibrosis[J]. Fibrogenesis Tissue Repair, 2012, 5(1): 1-9.
[8] Zeng CX, Wang YL, Xie C, et al. Identification of a novel TGF-β-miR-122-fibronectin 1/serum response factor signaling cascade and its implication in hepatic fibrogenesis[J]. Oncotarget, 2015, 6(14): 12224-12233.
[9] Chen C, Wu CQ, Zhang ZQ, et al. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation[J]. Exp Cell Res, 2011, 317(12): 1714-1725.
[10] Li WQ, Chen C, Xu MD, et al. The rno-miR-34 family is upregulated and targets ACSL1 in dimethylnitrosamine-induced hepatic fibrosis in rats[J]. FEBS J, 2011, 278(9): 1522-1532.
[11] Chen L, Charrier A, Zhou Y, et al. Epigenetic regulation of connective tissue growth factor by microRNA-214 delivery in exosomes from mouse or human hepatic stellate cells[J]. Hepatology, 2014, 59(3): 1118-1129.
[12] Lee CT, Risom T, Strauss WM. Evolutionary conservation of microRNA regulatory circuits: an examination of microRNA gene complexity and conserved microRNA-target interactions through metazoan phylogeny[J]. DNA Cell Biol, 2007, 26(4): 209-218.
[13] Friedman RC, Farh KKH, Burge CB, et al. Most mammalian mRNAs are conserved targets of microRNAs[J]. Genome Res, 2009, 19(1): 92-105.
[14] Lando D. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor[J]. Genes Dev, 2002, 16(12): 1466-1471.
[15] Csak T, Bala SS, Lippai D, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in diet-induced steatohepatitis[J]. Liver Int, 2015, 35(2): 532-541.
[16] Han J, He YP, Zhao H, et al. Hypoxia inducible factor-1 promotes liver fibrosis in nonalcoholic fatty liver disease by activating PTEN/p65 signaling pathway[J]. J Cell Biochem, 2019, 120(9): 14735-14744.
[17] Triantafyllou EA, Mylonis I, Simos G, et al. Hypoxia induces pro-fibrotic and fibrosis marker genes in hepatocellular carcinoma cells independently of inflammatory stimulation and the NF-κΒ pathway[J]. Hypoxia Auckl N Z, 2019, 7: 87-91.
[18] Zhang LT, Zhou D, Li JF, et al. Effects of bone marrow-derived mesenchymal stem cells on hypoxia and the transforming growth factor beta 1 (TGFβ-1) and SMADs pathway in a mouse model of cirrhosis[J]. Med Sci Monit, 2019, 25: 7182-7190.
[19] Ankoma-Sey V, Wang Y, Dai ZH. Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells[J]. Hepatology, 2000, 31(1): 141-148.
[20] Novo E, Cannito S, Zamara E, et al. Proangiogenic cytokines as hypoxiadependent factors stimulating migration of human hepatic stellate cells[J]. Am J Pathol, 2007, 170(6): 1942-1953.
[21] Zhan L, Huang C, Meng XM, et al. Hypoxia-inducible factor-1α in hepatic fibrosis: a promising therapeutic target[J]. Biochimie, 2015, 108: 1-7.
[22] Choi JH, Kim SM, Lee GH, et al. Platyconic acid A, Platycodi radix-derived saponin, suppresses TGF-β1-induced activation of hepatic stellate cells via blocking SMAD and activating the PPARγ signaling pathway[J]. Cells, 2019, 8(12): 1544.
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
天津医科大学学报(2021年4期)2021-08-21 02:14:50
中日友好医院学报(2021年1期)2021-04-14 01:58:32
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
山东医药(2020年9期)2020-05-20 01:12:16
解放军医学院学报(2020年12期)2020-03-29 05:11:32
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
中成药(2018年9期)2018-10-09 07:18:32
中国医药导报(2015年27期)2015-02-28 22:08:01
中华皮肤科杂志(2014年3期)2014-12-19 12:54:50
