
利拉鲁肽作用肠道固有淋巴细胞改善小鼠炎症性肠病
2020-08-18 10:05孙寒晓李咏梅盛慧明
上海交通大学学报(医学版) 2020年6期
李 月 ,孙寒晓,殳 洁,李咏梅,盛慧明#
1. 北华大学医学院,吉林132013;2. 上海交通大学医学院附属同仁医院检验科,上海200336
炎症性肠病(inflammatory bowel disease,IBD)是一种慢性炎症性肠道疾病,包括溃疡性结肠炎(ulcerative colitis,UC)和克罗恩病(Crohn's disease,CD)[1]。流行病学显示IBD 发病率在亚洲国家和其他发展中国家呈逐年上升趋势[2],严重影响患者的生活质量。目前,其发病机制尚未完全明确[3],临床上并无完全治愈IBD 的有效治疗手段。因此,进一步探讨IBD 的发病机制,寻找新的防治策略和干预靶点是目前亟待解决的关键问题。
胰高血糖素样肽-1 受体激动剂(glucagon-like peptide-1 receptor agonist,GLP-1RA)是目前治疗2 型糖尿病的有效药物,且经FDA 批准使用的GLP-1RA 主要有利拉鲁肽、艾塞那肽等[4]。研究[5]表明利拉鲁肽不仅具有血糖调节作用,还具有抗炎等特性,但关于利拉鲁肽参与肠道炎症的抗炎作用研究甚少。
随着对IBD 的作用机制的深入研究,人们逐渐聚焦到了一类新的免疫细胞——固有淋巴细胞(innate lymphoid cell,ILC)。ILC 是一类与T 淋巴细胞表型和功能相似的天然免疫细胞[6]。ILC 家族主要分为3 个亚群,分别是 1 型ILC(group 1 ILC,ILC1)和自然杀伤细胞(natural killer cell,NK) 细 胞、2 型ILC(group 2 ILC,ILC2)、淋巴组织诱导细胞和3 型ILC(group 3 ILC,ILC3)[7-8]。ILC 可分泌细胞因子,主要通过与间质细胞以及其他免疫细胞之间的相互作用在IBD 等多种自身免疫性疾病中发挥效应[9]。研究[10-11]显示ILC 参与肠道黏膜稳态的调节,并在IBD 的发生和发展中起着重要的作用。
葡聚糖硫酸钠盐(dextran sulfate sodium,DSS)诱导的小鼠结肠炎模型所致临床和病理特征与人类IBD 相似,因此是研究IBD 常用的动物疾病模型[12-13]。本研究通过对C57BL/6 小鼠进行DSS 诱导构建小鼠IBD 模型,应用利拉鲁肽进行治疗,从小鼠的粪便性状、结肠外观和病理改变、免疫细胞数量以及炎性细胞因子的表达等方面评估利拉鲁肽对DSS 诱导IBD 小鼠的作用,以期为辅助治疗IBD 提供新的理论和实验依据。
1 对象与方法
1.1 研究对象
1.1.1 实验动物 6 ~10 周龄C57BL/6 野生型雌性小鼠,体质量16 ~22 g,购于上海斯莱克实验动物有限责任公司,实验动物生产许可证号为SCXK(沪)2017-0005。所有小鼠均饲养在中国科学院上海生命科学研究所SPF 级动物房,实验动物使用许可证号为(沪)SYXK2019-0001。饲养条件如下:动物房室内温度18 ~22 ℃,相对湿度50%~60%,分笼饲养,自由摄食饮水,适应环境后进行实验。动物实验均得到中国科学院上海生命科学研究所医学研究伦理委员会批准。
1.1.2 实验仪器 GalliosTM流式细胞仪(美国贝克曼公司),ECLIPSE 80i 显微镜(日本尼康公司),ZHWY-100H经典型多振幅轨道摇床(中国智城分析仪器制造有限公司),Eppendorf AG 22331 Hamburg 低速离心机(德国艾本德公司)。
1.1.3 实验试剂 FITC 标记的CD3e 抗体、CD11b 抗体、CD11c 抗体、B220 抗体、粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colony stimulating factor,GMCSF)抗体,PE 标记的白介素-5 (interleukin-5,IL-5)抗体,APC 标记的视黄酸相关的孤儿受体γt (retinoid-related orphan receptor γt,RORγt) 抗 体,PerCP-Cy5.5 标 记 的CD11b 抗体、IL-17A 抗体,PE-Cy7 标记的IL-13 抗体、CD3e 抗体、CD11b 抗体(美国eBioscience 公司);PE 标记的Siglec-F(sialic acid-binding immunoglobulin-like lectin F)抗体,APC 标记的GATA3(GATA binding protein 3)抗 体、Ly6G(lymphocyte antigen 6 complex locus protein G6D)抗体(美国BD Biosciences 公司);PE 标记的IL-22抗体,PE-Cy7 标记的CD11c 抗体,CD16/32 抗体(美国Biolegend 公司);PE-Cy7 标记的B220 抗体(美国Thermo Fisher Scientific 公 司 )。PMA(phorbol-12-myristate-13-acetate)(美国Sigma 公司,货号H9268-10G),离子霉素(美国Sigma 公司,货号10004974-1),布雷菲德菌素A(美国Sigma 公司,货号11861-5),LIVE/DEAD®固定式死细胞染色试剂盒(美国Invitrogen 公司,货号L34955),Foxp3/转录因子染色缓冲液(美国Thermo Fisher Scientific公司,货号00-5523-00),苏木精- 伊红(hematoxylineosin,H-E)染色试剂盒(中国碧云天公司,货号C0105),DSS(美国MP Biomedicals 公司,货号160110),利拉鲁肽诺和力注射液(丹麦诺和诺德公司,规格18 mg/3 mL),二硫苏糖醇(美国Promega 公司,货号V3155),乙二胺四乙酸(美国Invitrogen 公司,货号25200-072),胎牛血清(美国GEMINI 公司,货号900-108),脱氧核糖核酸酶Ⅰ(美国Sigma 公司,货号DN25-1G),胶原酶Ⅷ(美国Sigma 公司,货号C2139-500M),RPMI 1640 培养基(美国Invitrogen 公司,货号22409015),Percoll 细胞分离液(德国Amersham/GE 公司,货号17089102)。
1.2 实验方法
1.2.1 动物分组及模型构建 适应性喂养7 d 后,随机选取20 只小鼠分为4 组,每组5 只,分别为对照组、利拉鲁肽组、模型组和治疗组。对照组小鼠每日饮用无菌水,并腹腔注射磷酸盐缓冲液(phosphate buffer saline,PBS);利拉鲁肽组小鼠每日饮用无菌水,并腹腔注射利拉鲁肽;模型组小鼠每日饮用DSS 浓度为3%的无菌水溶液,并腹腔注射PBS;治疗组小鼠每日饮用DSS 浓度为3%的无菌水溶液,并腹腔注射利拉鲁肽。注射液现用现配,PBS 剂量0.6 mg/ (kg·d),利拉鲁肽剂量0.6 mg/ (kg·d),共处理7 d。每日观察并记录小鼠体质量、粪便形态以及有无便血等情况,判断结肠炎严重程度。
1.2.2 小鼠肠道固有层细胞的提取 无菌环境下取小鼠结肠,PBS 洗涤后,室温下在含有1 mmol/L 二硫苏糖醇的PBS 中振摇10 min,经PBS 洗涤,在含有30 mmol/L 乙二胺四乙酸的PBS 中于37 ℃振荡孵育10 min,重复 2 次。加入含有1% 胎牛血清、脱氧核糖核酸酶Ⅰ和胶原酶Ⅷ的RPMI 1640 培养液,在37 ℃、5% CO2的培养箱消化1.5 h 后,剧烈摇动组织匀浆,使其通过100 µm 细胞过滤器,室温以1 048×g 离心20 min 后,从80%和40% Percoll 梯度液的中间层收获肠道固有层细胞。
1.2.3 流式细胞术分析 肠道固有层谱系(lineage,Lin)标志物包括CD3e、B220、CD11b 和CD11c。应用流式细胞术检测小鼠结肠内的中性粒细胞(CD11b+Ly6G+)、嗜酸 性 粒 细 胞(CD11b+Siglec-F+)、ILC2(Lin-GATA3+)和ILC3(Lin-RORγt+),以评估肠炎的严重程度。将分离出的肠道固有层细胞,用50 ng/mL PMA 和500 ng/mL 离子霉素刺激4 h,在收获细胞前2 h 添加2 µg/mL 布雷菲德菌素A。收集细胞,使用CD16/32 抗体室温孵育5 min,然后稀释CD3e 抗体、B220 抗体、CD11b 抗体、CD11c抗体、Siglec-F 抗体和Ly6G 抗体,4 ℃孵育30 min 进行细胞表面蛋白的染色。经PBS 洗涤后,根据说明书使用Foxp3/转录因子染色缓冲液将细胞进行固定透化,胞内转录因子(GATA3、 RORγt)、细胞因子(IL-5、 IL-13、IL-17、IL-22 和GM-CSF) 于4 ℃孵 育45 min,PBS 洗涤重悬后经GalliosTM流式细胞仪检测分析,数据经Flow JoV10 软件分析。
1.2.4 组织学分析 取小鼠肛门以上1 cm 处的结肠组织,长约0.5 cm,PBS 洗涤后用4%多聚甲醛固定24 h,将组织包埋在石蜡中,制备5 µm 厚度的组织切片,进行H-E 染色。使用光学显微镜随机选取3 个视野,观察结肠的病理改变。根据黏膜层炎症细胞浸润程度(1 ~3 分)、肌层炎症细胞浸润程度(0 ~2 分)、杯状细胞缺失情况(0 ~2 分)、黏膜糜烂至溃疡程度(0 ~2 分)等4 个方面,采用盲法进行组织病理学分析并评分,最高得分为 9 分[14]。
1.3 统计学方法
采用GraphPad Prism 6.0 软件进行统计学分析,定量资料用±s 表示,组间比较采用非配对Student's t 检验。P<0.05 时认为差异具有统计学意义。
2 结果
2.1 利拉鲁肽显著缓解DSS 诱导的IBD 小鼠结肠损伤
实验第5 日,模型组小鼠体质量较第1 日下降12.23%,第6 ~7 日体质量继续下降。实验过程中观察到小鼠有稀便甚至血便现象,视为建模成功。模型组小鼠肠道内血便情况较为严重,而治疗组血便情况相对较轻。实验第5 日,与对照组相比,模型组小鼠体质量明显降低(P=0.014);处理7 d 后,与模型组相比,治疗组小鼠体质量下降明显缓解(P=0.043)(图1A)。小鼠结肠长度比较结果显示,模型组小鼠结肠长度明显短于对照组(P=0.008);而治疗组与模型组相比,结肠缩短情况明显缓解(P=0.007)(图1B、C)。
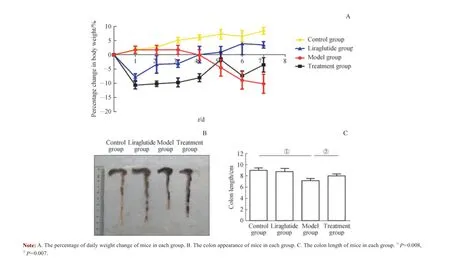
图1 利拉鲁肽显著缓解DSS 诱导的IBD 小鼠结肠损伤Fig 1 Liraglutide significantly alleviated colon injury induced by DSS in IBD mice
2.2 利拉鲁肽显著缓解DSS 诱导的IBD 小鼠结肠黏膜屏障损伤
H-E 染色结果显示,模型组小鼠黏膜上皮细胞大量坏死脱落,杯状细胞缺失,黏膜下层存在淋巴细胞重度浸润,几乎没有完整的黏膜结构(图2A)。而经过利拉鲁肽处理后,组织学损伤得到了明显改善,结肠组织结构、固有层肠腺、黏膜肌层相对完整,黏膜下层轻中度淋巴细胞浸润。组织病理学评分显示,与对照组相比,模型组小鼠评分明显升高(P=0.008);而经过利拉鲁肽处理后,评分明显降低(P=0.008)(图2B)。

图2 利拉鲁肽显著缓解DSS 诱导的IBD 小鼠结肠黏膜屏障损伤Fig 2 Liraglutide significantly alleviated the colonic mucosal barrier injury induced by DSS in IBD mice
2.3 利拉鲁肽改善DSS 诱导的IBD 小鼠结肠内炎症细胞浸润
应用流式细胞术检测小鼠结肠内嗜酸性粒细胞和中性粒细胞比例,以评估肠炎的严重程度,其分选策略如图3A所示。结果(图3B ~E)显示模型组小鼠结肠内嗜酸性粒细胞、中性粒细胞比例明显高于对照组(均P=0.001),经过利拉鲁肽处理后,中性粒细胞、嗜酸性粒细胞比例较模型组显著降低(P=0.004,P=0.002),说明利拉鲁肽能够改善DSS 诱导的IBD 小鼠结肠内炎症细胞的浸润。
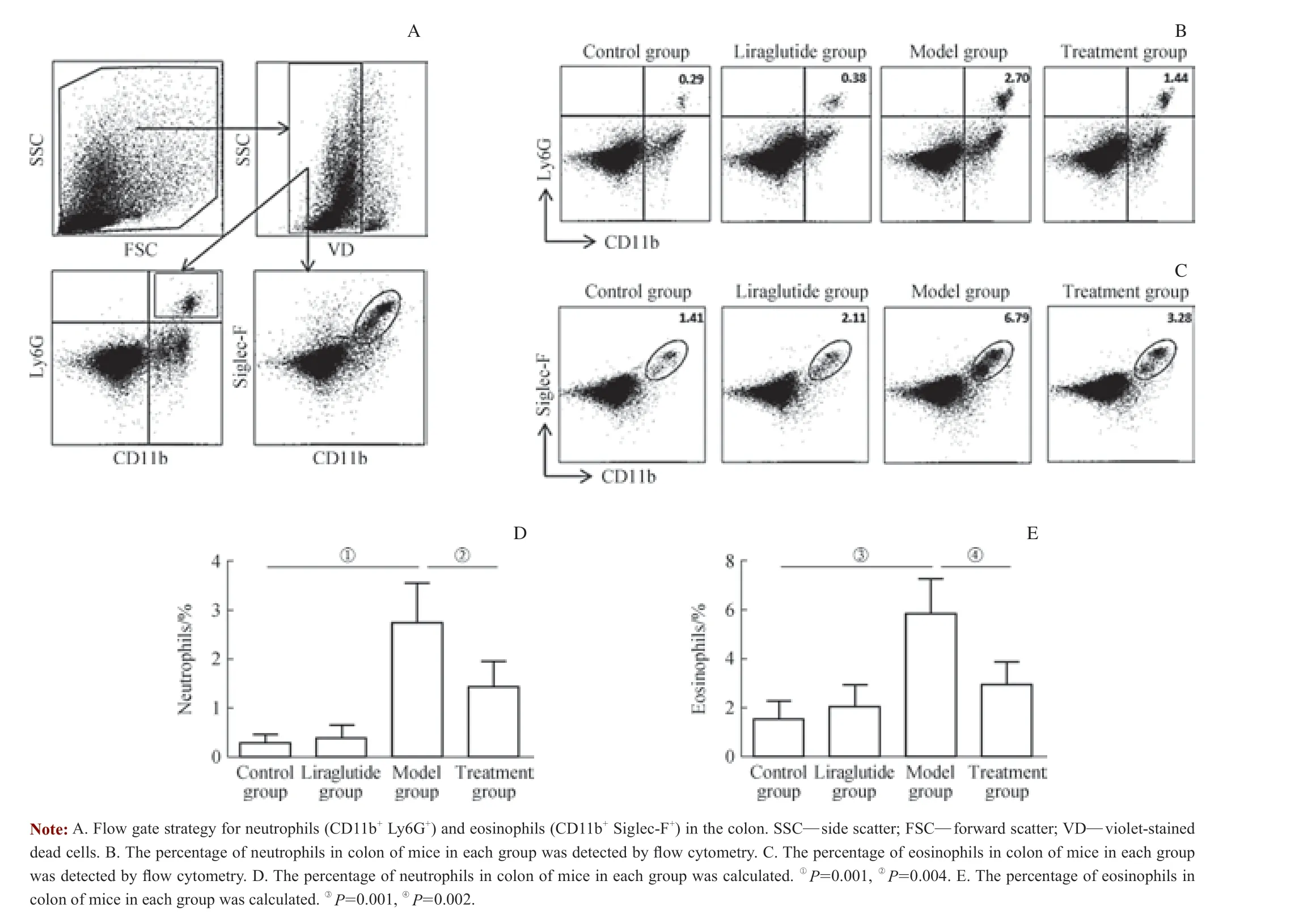
图3 利拉鲁肽改善DSS 诱导的IBD 小鼠结肠内炎症细胞浸润Fig 3 Liraglutide improved the colonic inflammatory cell infiltration induced by DSS in IBD mice
2.4 利拉鲁肽影响DSS 诱导的IBD 小鼠肠道ILC 的变化
应用流式细胞术检测小鼠结肠ILC2、ILC3 及其分泌的细胞因子(分选策略如图4A、B 所示),评估利拉鲁肽处理前后ILC2、ILC3 数量和功能的变化。结果显示,与对照组相比,模型组小鼠结肠内ILC2 占总ILC 的比例升高(P=0.032),而经过利拉鲁肽处理后ILC2 比例较模型组降低(P=0.032),提示利拉鲁肽可抑制结肠内ILC2 的数量(图4C);但通过比较模型组与治疗组之间ILC2 分泌的主要细胞因子IL-5、IL-13、IL-17 的水平,未发现差异有统计学意义(结果未显示)。与模型组相比,治疗组ILC3 的比例明显增加(P=0.008)(图4D),其IL-22 的分泌水平亦明显升高(P=0.008)(图4E),但2 组间其他细胞因子IL-17、GM-CSF 分泌水平的差异无统计学意义(结果未显示)。
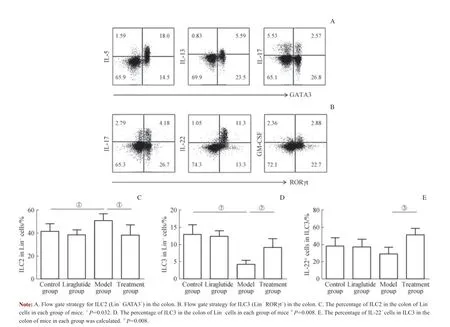
图4 利拉鲁肽影响DSS 诱导的IBD 小鼠肠道ILC 的变化Fig 4 Liraglutide affected the intestinal ILC of IBD mice induced by DSS
3 讨论
本研究通过分析小鼠的体质量变化、结肠长度以及结肠的病理学改变、免疫细胞数量,明确利拉鲁肽可延缓DSS 诱导的小鼠IBD 的发生发展。对结肠内ILC 及其分泌的细胞因子分析后发现,经利拉鲁肽处理后,DSS 诱导的IBD 小鼠结肠内ILC2 比例明显降低,ILC3 比例明显升高,且ILC3 分泌的细胞因子IL-22 的水平显著增加。
ILC3 主要存在于肠黏膜组织中,表达转录因子RORγt, 主要分泌IL-22、IL-17 和GM-CSF 等细胞因子,在黏膜稳态和炎症反应中起重要作用[15]。IL-22 信号会诱导黏蛋白产生,还可通过促进上皮细胞的增殖和存活来促进组织修复。此外,IL-22 还能促进核苷酸寡聚结构域蛋白2(nucleotide oligomerization domain-containing protein 2,NOD2)产生,而NOD2 信号的激活也可促进黏蛋白分泌,保护肠上皮细胞免受细菌侵袭。ILC3 产生的IL-22 可与肠上皮细胞表达的受体结合,通过激活蛋白酪氨酸激酶-信号转导子与转录激活子(just another kinase-signal transducer and activator of transcription,JAK-STAT)信号通路,促进上皮细胞增殖,从而使上皮屏障保持完整性[16]。同时,IL-22 可以诱导产生抗微生物因子Reg( regenerating) - Ⅲβ 和Reg- Ⅲγ,限制共生菌与上皮细胞接触,避免肠道炎症产生[17]。在本研究中,利拉鲁肽作用后,肠固有层ILC3 比例及IL-22 的分泌明显增加,说明IL-22 可能通过以上方式在肠道黏膜免疫系统的调节中发挥重要作用。
ILC2 主要分布在肠道、肺部、淋巴组织、肝脏和肠系膜中[18],主要分泌Ⅱ型细胞因子如IL-4、IL-5 和IL-13等。GATA3 是ILC2 的主要转录因子[19],是ILC2 发育、存活和功能发挥的关键调控因子[20]。在唑酮诱导的小鼠结肠炎模型中,ILC2 由于受到IL-25 的刺激,可加速促进肠道炎症[21]。在新确诊的IBD 患者中,疾病早期病变处肠黏膜内ILC2 比例明显增加[22],提示ILC2 可能在IBD中发挥促进炎症的作用。另有研究[23]发现GLP-1RA 可作用于自身免疫性疾病,如抑制IL-33 的释放以及过敏气道炎症,推测GLP-1RA 可能是治疗ILC2 介导的过敏性哮喘的一种新手段。结合本研究结果,GLP-1RA 利拉鲁肽可能通过抑制肠道ILC2 从而改善DSS 诱导的小鼠IBD,但其具体机制尚需进一步研究。
近年来研究[24]发现,ILC 与辅助性T 细胞类似,具有一定的可塑性。ILC2 在发育和维持过程中可能向ILC3 转化,炎性ILC2 可能分化产生IL-17 的ILC3 样细胞[25]。在人类CD 患者的肠样本中发现ILC2 分泌的IL-13 和γ- 干扰素(interferon-γ,IFN-γ)明显增加[26],说明炎性ILC3 可能转变为分泌IFN-γ 的ILC2 样细胞。同时本实验中ILC2比例降低伴ILC3 比例升高也提示,存在ILC2 向ILC3 转化的可能性。既往研究表明ILC2 分泌的IL-4 对于抵抗细胞外寄生虫感染的保护性免疫至关重要[27],同时也与IBD的严重程度有关[28]。所以利拉鲁肽是否影响ILC2 分泌IL-4,进而减缓疾病的进程仍需后续研究进一步明确。
此外,本研究仍具有一定的局限性:首先,DSS 诱导的IBD 小鼠模型仅模仿了UC 患者一些典型的组织病理学特征和某些免疫学特征,并不能完全代表CD 的复杂特征;其次,样本例数较少。因此,迫切需要开发能够解决现有局限性且更接近CD 和UC 特征的其他实验模型[29],并增加研究病例数以明确实验结论。
综上,本研究发现GLP-1RA 利拉鲁肽可能作用于肠道ILC,明显延缓DSS 诱导的IBD 小鼠的疾病发生与发展,为利拉鲁肽的潜在治疗用途提供了理论依据。
参·考·文·献
[1] Bian XY, Wu WR, Yang LY, et al. Administration of Akkermansia muciniphila ameliorates dextran sulfate sodium-induced ulcerative colitis in mice[J]. Front Microbiol, 2019, 10: 2259.
[2] Parada Venegas D, De la Fuente MK, Landskron G, et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases[J]. Front Immunol, 2019, 10: 277.
[3] Nie Y, Lin QL, Luo FJ. Effects of non-starch polysaccharides on inflammatory bowel disease[J]. Int J Mol Sci, 2017, 18(7): 1372.
[4] Trujillo JM, Nuffer W, Ellis SL. GLP-1 receptor agonists: a review of head-tohead clinical studies[J]. Ther Adv Endocrinol Metab, 2015, 6(1): 19-28.
[5] Yaribeygi H, Maleki M, Sathyapalan T, et al. Anti-inflammatory potentials of incretin-based therapies used in the management of diabetes[J]. Life Sci, 2020, 241: 117152.
[6] Lim AI, Di Santo JP. ILC-poiesis: ensuring tissue ILC differentiation at the right place and time[J]. Eur J Immunol, 2019, 49(1): 11-18.
[7] Putignani L, Del Chierico F, Vernocchi P, et al. Gut microbiota dysbiosis as risk and premorbid factors of IBD and IBS along the childhood-adulthood transition[J]. Inflamm Bowel Dis, 2016, 22(2): 487-504.
[8] Lim AI, Verrier T, Vosshenrich CA, et al. Developmental options and functional plasticity of innate lymphoid cells[J]. Curr Opin Immunol, 2017, 44: 61-68.
[9] Ebbo M, Crinier A, Vély F, et al. Innate lymphoid cells: major players in inflammatory diseases[J]. Nat Rev Immunol, 2017, 17(11): 665-678.
[10] Marion-Letellier R, Savoye G, Ghosh S. IBD: in food we trust[J]. J Crohns Colitis, 2016, 10(11): 1351-1361.
[11] Forkel M, Mjösberg J. Dysregulation of group 3 innate lymphoid cells in the pathogenesis of inflammatory bowel disease[J]. Curr Allergy Asthma Rep, 2016, 16(10): 73.
[12] Mizoguchi A. Animal models of inflammatory bowel disease[J]. Prog Mol Biol Transl Sci, 2012, 105: 263-320.
[13] Eichele DD, Kharbanda KK. Dextran sodium sulfate colitis murine model: an indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis[J]. World J Gastroenterol, 2017, 23(33): 6016-6029.
[14] Pavlick KP, Ostanin DV, Furr KL, et al. Role of T-cell-associated lymphocyte function-associated antigen-1 in the pathogenesis of experimental colitis[J]. Int Immunol, 2006, 18(2): 389-398.
[15] Zeng BN, Shi SN, Ashworth G, et al. ILC3 function as a double-edged sword in inflammatory bowel diseases[J]. Cell Death Dis, 2019, 10(4): 315.
[16] Guo XH, Qiu J, Tu T, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection[J]. Immunity, 2014, 40(1): 25-39.
[17] Xu X, Fukui H, Ran Y, et al. The link between type Ⅲ Reg and STAT3-associated cytokines in inflamed colonic tissues[J]. Mediators Inflamm, 2019, 2019: 7859460.
[18] Peters CP, Mjösberg JM, Bernink JH, et al. Innate lymphoid cells in inflammatory bowel diseases[J]. Immunol Lett, 2016, 172: 124-131.
[19] Halim TY, MacLaren A, Romanish MT, et al. Retinoic-acid-receptor-related orphan nuclear receptor α is required for natural helper cell development and allergic inflammation[J]. Immunity, 2012, 37(3): 463-474.
[20] Hoyler T, Klose CS, Souabni A, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells[J]. Immunity, 2012, 37(4): 634-648.
[21] Camelo A, Barlow JL, Drynan LF, et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13[J]. J Gastroenterol, 2012, 47(11): 1198-1211.
[22] Forkel M, van Tol S, Höög C, et al. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn's disease and ulcerative colitis[J]. J Crohns Colitis, 2019, 13(1): 67-78.
[23] Toki S, Goleniewska K, Reiss S, et al. Glucagon-like peptide 1 signaling inhibits allergen-induced lung IL-33 release and reduces group 2 innate lymphoid cell cytokine production in vivo[J]. J Allergy Clin Immunol, 2018, 142(5): 1515-1528. e8.
[24] Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity[J]. Immunity, 2018, 48(6): 1104-1117.
[25] Krabbendam L, Bal SM, Spits H, et al. New insights into the function, development, and plasticity of type 2 innate lymphoid cells[J]. Immunol Rev, 2018, 286(1): 74-85.
[26] Lim AI, Menegatti S, Bustamante J, et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells[J]. J Exp Med, 2016, 213(4): 569-583.
[27] Panda SK, Colonna M. Innate lymphoid cells in mucosal immunity[J]. Front Immunol, 2019, 10: 861.
[28] Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis[J]. J Clin Invest, 2004, 113(10): 1490-1497.
[29] Bamias G, Arseneau KO, Cominelli F. Mouse models of inflammatory bowel disease for investigating mucosal immunity in the intestine[J]. Curr Opin Gastroenterol, 2017, 33(6): 411-416.
猜你喜欢
西南医科大学学报(2022年2期)2022-11-23
福建医科大学学报(2022年3期)2022-09-10
中国药学药品知识仓库(2022年1期)2022-03-23
疯狂英语·新策略(2021年7期)2021-08-26
天津医科大学学报(2021年4期)2021-08-21
服饰导报·鞋世界(2021年4期)2021-05-17
实用中西医结合临床(2020年5期)2020-12-23
医食参考(2017年12期)2017-04-01
中国动物保健(2015年4期)2015-10-21
网球俱乐部(2009年5期)2009-06-15
