
柱芳烃与稀有气体分子间相互作用的理论研究*
2020-08-12 11:09孙涛
广州化工 2020年14期
孙 涛
(贵州大学网络与信息化管理中心,贵州省高性能计算化学重点实验室,贵州 贵阳 550025)
最近几年,稀有气体(Ng)主客体复合物发展迅速,许多研究者已经从理论计算和实验上研究稀有气体的主客体复合物。Frenking等[1]通过理论计算发现在C60内,Xe-Xe 能够形成共价键,而He-He 和Ne-Ne之间为弱非键相互作用。Chattaraj等使用密度泛函(DFT)方法,从结合能、解离能和焓变研究了六元瓜环(cucurbit[6]uril,CB[6])与Ne、Ar、Kr分子间相互作用,结果表明CB[6]可以与3个Ne原子形成主客体复合物,经零点能校正后,解离能在3.4~4.1 kcal/mol之间;而CB[6]只能与1个Ar或1个Kr形成复合物[2]。柱芳烃是冠醚、环糊精、杯芳烃和葫芦脲后一类新型的大环化合物,由于其结构独特和优异的主客体化学已逐渐成为超分子化学研究和发展的热点之一[3]。
稀有气体与C6H6之间存在弱的非键相互作用[4],而甲基柱[5]芳烃5个苯环单元,甲基柱[5]芳烃的静电势如图1所示,分子静电势图能更直观地反映出原子荷电的情况,其红色区域表示负电区,蓝色区域表示正电区,电子聚集在柱芳烃腔内苯环和端基氧区域。柱芳烃与稀有气体能否以非键相互作用形成主客体复合物,如果二者之间能形成主客体复合物,结合能是多大?二者的相互作用是静电占主导,还是色散占主导?因此本文应用量子化学计算方法研究MeP5…Ng(Ng=He、Ne、Ar、Kr、Xe)复合物分子间非键相互作用,计算其结合能,并分析其相互作用本质。
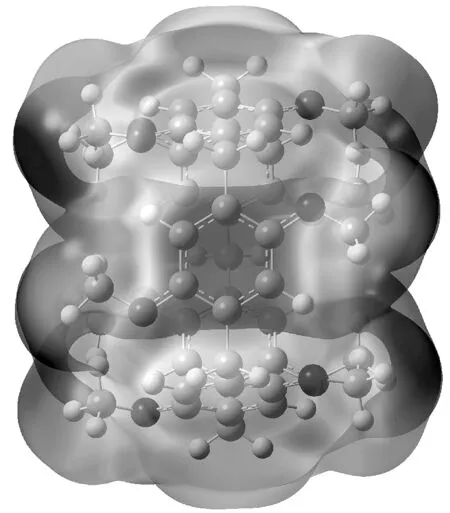
图1 MeP5分子静电势图
1 计算方法
Head-Gordon发展的杂化泛函ωB97X-V[5]在计算分子间非键相互作用结果较准确,为了验证该方法是否适合计算MeP5…Ng(Ng=He、Ne、Ar、Kr、Xe)。将MeP5…Ar复合物的柱芳烃进行裁剪,如图2所示。首先垂直移动Ar,距离C10H14O2苯环平面在3.2~4.0 Å之间;找到Ar距C10H14O2的平面稳定距离为3.4 Å,保持该距离,水平向外移动Ar。使用ωB97X-V/def2-svpd[6]计算C10H14O2…Ar复合物的结合能,并与金标准的CCSD(T)/CBS(计算方法如公式1) 作比较,探讨ωB97X-V的可靠性。C10H14O2…Ar复合物计算结果如图3所示。
(1)
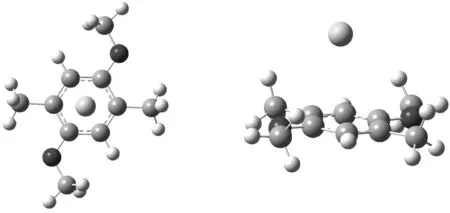
图2 C10H14O2…Ar 俯视和侧面图
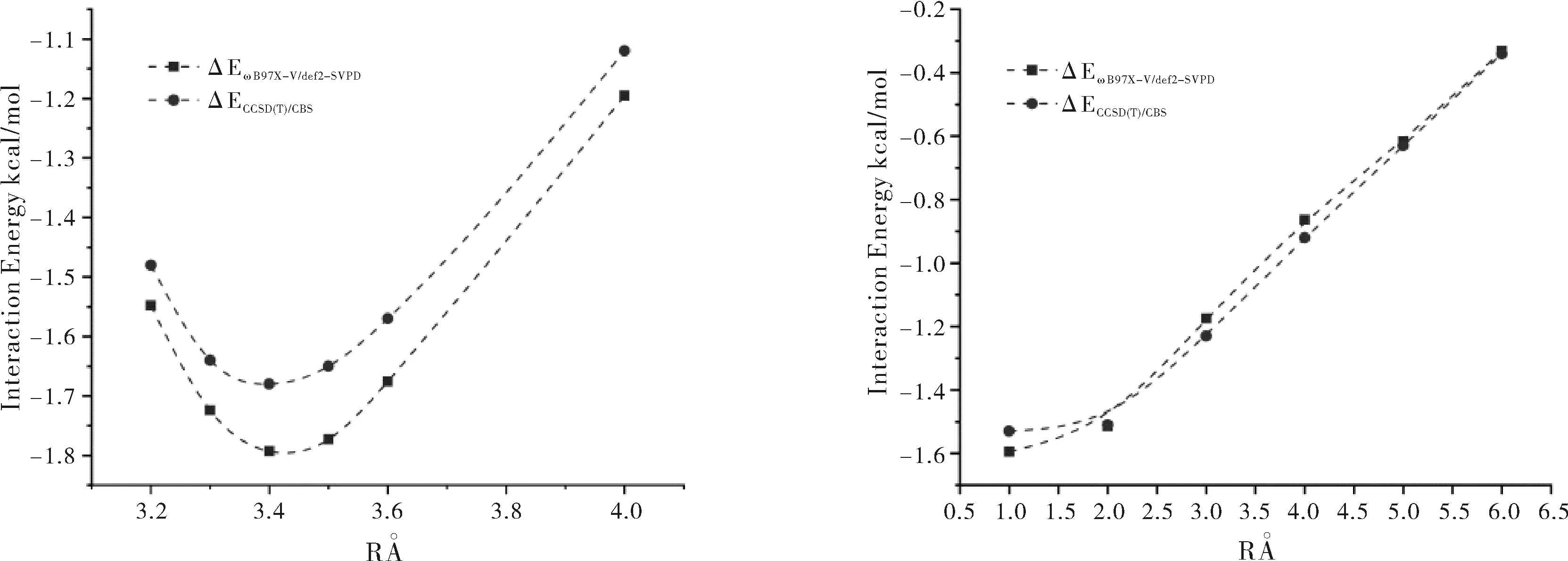
图3 ωB97X-V/def2-svpd与CCSD(T)/CBS的计算结果
由图3可以看出,ωB97X-V/def2-SVPD与CCSD(T)/CBS计算结果相差不大,而且变化趋势也一致,因此使用ωB97X-V/def2-SVPD进行几何结构和结合能计算。使用Boys和Bernardi完全均衡校正法[7]进行基函数重叠误差(BSSE)校正后计算其结合能(E)。为了探讨其相互作用本质,使用ALMO-EDA(Energy decomposition analysis based on absolutely-localized molecular orbitals)方法[8-9],对MeP5…Ng(Ng=He, Ne, Ar, Kr, Xe)分子间相互作用进行能量分解分析,将结合能分解为“冻结密度”项EFRZ、极化项EPol和电荷转移项ECT。冻结能量项进一步分解为静电能EElec、Pauli排斥EPauli和色散能EDisp。所有的计算工作在贵州省高性能计算化学重点实验室和贵州大学云计算平台完成。
2 结果与讨论
2.1 几何结构和结合能
在ωB97X-V/def2-SVPD水平,对MeP5…Ng(Ng=He, Ne, Ar, Kr, Xe)体系的复合物几何结构进行优化,得到平衡结构如图4所示,在MeP5…Ng(Ng=He, Ne, Ar, Kr, Xe)体系的复合物中,稀有气体在柱芳烃的空腔内,稀有气体Ng到柱芳烃MeP5底部平面的距离在3.09~4.04 Å之间,随着原子序数的增加,R逐渐变长,到Kr后不再改变,这是因为Kr,Xe均在MeP5的中心点,二者均为4.04 Å。复合物中分子间主要存在众多的Ng…π相互作用,结合能在-1.01~-11.20 kcal/mol,其相互作用能远大于C6H6…Ng(Ng=He, Ne, Ar, Kr, Xe)[4]。Ng(Ng=He, Ne)原子半径较小,MeP5空腔半径较大,在MeP5空腔内偏向一侧,结合能也相对较小,理论计算表明MeP5还可以容纳两个Ng(Ng=He, Ne)原子。
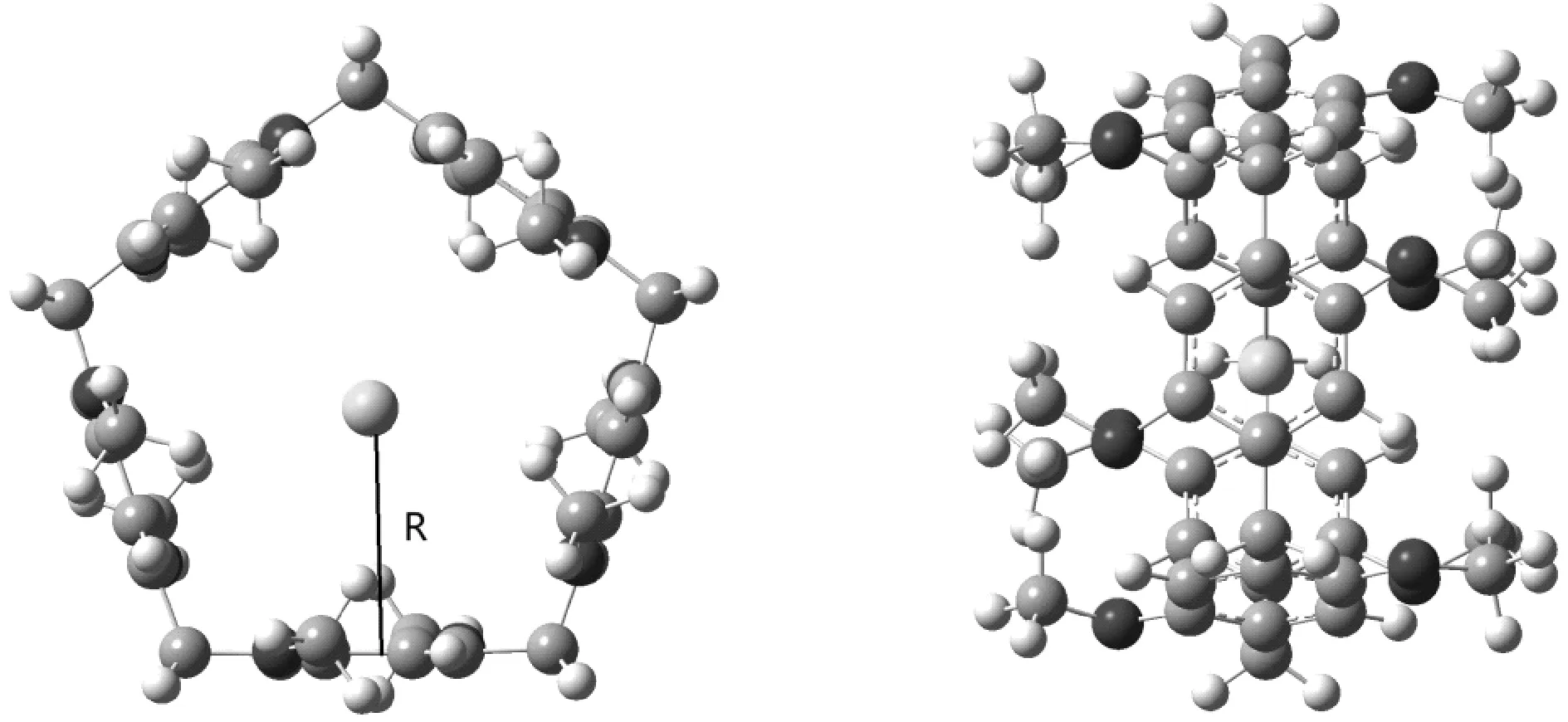
图4 MeP5…Ng(Ng=He、Ne、Ar、Kr、Xe)的俯视和侧面图
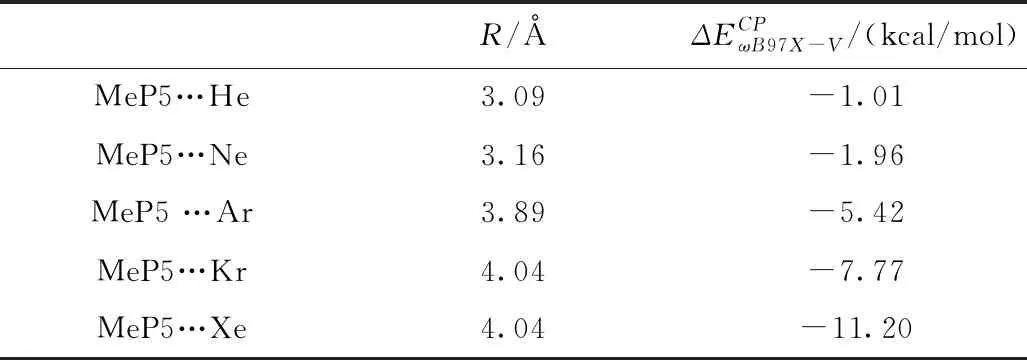
表1 MeP5…Ng(Ng= He, Ne, Ar, Kr, Xe)的结合能
2.2 ALMO-EDA计算
使用ALMO-EDA对MeP5…Ng(Ng= He, Ne, Ar, Kr, Xe)的结合能进行能量分解分析,结果见表2,因Xe使用了ECP,目前还不能得到静电项和Pauli排斥。ALMO-EDA计算结果表明EElec、EDisp、EPol和ECT(MeP5…Ne为排斥作用)都是吸引作用,而Pauli排斥能EPauli是排斥作用,对于MeP5…Ng(Ng=He, Ne, Ar, Kr, Xe)复合物,色散能起主导作用,约占总吸引作用的57%~73%,极化项和电荷转移能约占总吸引相互作用的3%左右(MeP5…He约为11%)。随着原子序数的增加,色散能所占比例也有递增的趋势。在能量分解项中,色散能远大于静电能,起主导作用。
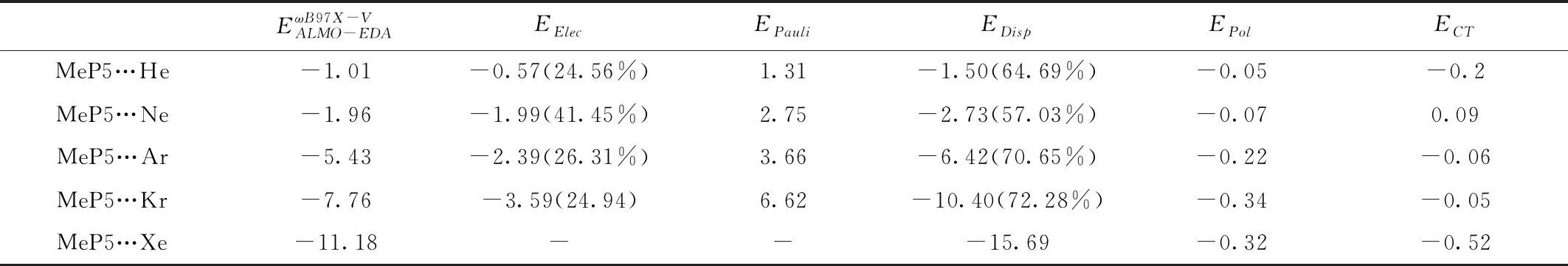
表2 MeP5…Ng(Ng= He, Ne, Ar, Kr, Xe)复合物ALMO-EDA相互作用能及其各作用项
3 结 语
密度泛函理论ωB97X-V/def2-SVPD计算MeP5…Ng(Ng=He、Ne、Ar、Kr、Xe)复合物的几何结构和结合能,结果表明,ωB97X-V/def2-SVPD方法MeP5…Ng的计算结果是准确的,在MeP5空腔内与Ng(Ng=He、Ne、Ar、Kr、Xe)主要存在Ng…π相互作用;随着原子序数的增加,结合能逐渐增大;二代ALMO-EDA能量分解分析结果表明,色散能是其主要吸引作用,起主导作用,而极化能和电荷转移能较小。
猜你喜欢
中学生数理化·八年级物理人教版(2022年10期)2022-11-10
大学物理(2022年9期)2022-09-28
中学生数理化·八年级物理人教版(2021年10期)2021-11-22
西藏艺术研究(2021年3期)2021-06-02
中学生数理化·八年级物理人教版(2020年11期)2020-12-14
马克思主义哲学研究(2020年2期)2020-07-21
物理通报(2020年7期)2020-07-01
中学生数理化·八年级物理人教版(2019年10期)2019-11-25
大观(2017年2期)2017-04-07
原子与分子物理学报(2015年3期)2015-11-24
