
相转移法合成纳米SAPO-34及其催化二甲醚制烯烃性能
2020-07-11 02:40张云峰樊卫斌朱卡克
化学反应工程与工艺 2020年5期
王 佳,张云峰,樊卫斌,朱卡克
1.华东理工大学,化学工程联合国家重点实验室,上海 200237;
2.中科院山西煤化所,山西 太原 030001
磷酸盐分子筛(SAPOs)是一类具有微孔的结晶固体,其独特的微孔结构和中强酸性赋予其良好的酸催化性能和择形选择性,在工业催化中得到广泛应用。其中,具有三维8元环(3.8 Å×3.8 Å)孔道的SAPO-34分子筛,由于其具有合适的中强度酸性以及只允许小分子通过的孔口和相对较大的笼(9.4 Å×12.7 Å),在甲醇或二甲醚转化制低碳烯烃(乙烯或丙烯)催化反应(MTO或DTO)中表现出高烯烃选择性[1-2],已成为工业MTO的首选催化剂。MTO作为非石油路线(煤或天然气)获取烯烃原料的重要途径,对我国减少国外石油依赖具有重要意义[3]。从已知反应机理看,MTO主要通过烯烃循环和芳烃循环机理生成烯烃,但反应进行的同时也会生成部分稠环芳烃,由于其体积较大,容易堵塞催化剂孔道,形成积炭,导致催化剂快速失活、缩短寿命[4]。目前,MTO催化剂设计的主要方向是在不影响低碳烯烃选择性的前提下延长催化剂寿命[5-6]。通过减小晶体尺寸至纳米级别或引入增强传质的多级孔道结构可以显著提高催化剂寿命,成为近年来分子筛领域研究的热点[7]。
为此,研究者尝试通过硬模板法[8-10]、软模板法[11-12]、干凝胶转化法[13-14]、微波/超声辅助结晶[15-16]、后处理[17-18]和无溶剂法[19-20]等合成纳米晶体或多级孔道结构的SAPO-34。迄今为止,只有微波辅助结晶[21]、干凝胶转化[13]和硅源缓释法[22]可以制备小于100 nm的SAPO-34晶体。但这些方法存在成本高、产率低、过程复杂或难以控制和酸强度不易受控等问题。
相转移法[23]是本课题组近来开发的一种制备纳米SAPO-34的新方法,主要是基于提高成核速率,增加原料在成核一步的消耗量,从而抑制其在晶化后期的生长;同时,相界面的存在也部分阻隔了颗粒之间的聚集生长(即非经典取向连接生长)。甲苯-水两相溶剂在动态晶化条件下,溶解于甲苯中的异丙醇铝快速转移至相界面,并与溶解于水中的磷源和硅源进行反应,提高了相界面处的浓度(过饱和度),从而引起爆发成核,快速消耗了大量的原料,使形成的晶核不能继续生长,相界面的阻隔作用在一定程度上避免了晶核的融合。与此不同,在普通水热合成SAPO过程中,往往先形成无定型的固体SAPO前驱体,后者在晶化过程中逐渐溶解,部分参与成核,因而成核速率慢,后续晶体生长也伴随着无定型SAPO前驱体的溶解,晶体容易长大。相转移合成法具有成核速率快、生长速率慢、产物晶体尺寸小、结晶度高、产物易分离和母液可回收利用等优点,是一种廉价和可放大的合成方法,但此前所使用的四乙基氢氧化铵(TEAOH)价格较高,增加了工业生产成本。为此,本工作在此前工作的基础上,以吗啡啉(MOR)和TEAOH作为共模板,采用相转移策略制备纳米尺度SAPO-34分子筛,期望在控制酸性和晶粒大小的情况下,可以降低合成成本。作为比较,采用水热法合成了常规SAPO-34分子筛,并以DTO作为探针反应,比较评价了二者的催化性能。
1 实验部分
1.1 催化剂制备
相转移合成纳米SAPO-34分子筛。原料SiO2、Al2O3、P2O5、TEAOH、MOR、H2O和甲苯物质的量之比为0.6:1.0:1.0:(y-x):x:50.0:6.5(其中y为共模板总量,可取值为2或3)。将3.84 g磷酸溶解于9.77 g二次去离子水中,滴加2.15 g正硅酸乙酯(TEOS),搅拌4 h至完全水解,然后按照配比依次加入TEAOH和MOR,室温下搅拌8 h,将此混合液标记为A;称取6.95 g异丙醇铝(TCI),在机械搅拌下缓慢溶解于10.00 g甲苯中,完全溶解后,标记为溶液B;将溶液A和B混合,转移至水热反应釜,在200 ℃动态晶化48 h,转速为50 r/min。晶化结束后,将得到的初级产物经洗涤,80 ℃干燥,600 ℃焙烧10 h后,标记为SAPO-34-TM-N,其中TM分别表示TEAOH和MOR。
水热法合成常规SAPO-34分子筛。原料SiO2,Al2O3,P2O5,TEAOH,MOR和H2O物质的量之比为0.6:1.0:1.0:1.02:1.98:50.0。将3.84 g磷酸溶解在9.77 g二次去离子水中,在剧烈搅拌过程中缓慢加入6.95 g异丙醇铝,搅拌6 h,然后滴加7.15 g正硅酸乙酯,搅拌4 h,最后加入2.92 g MOR,室温搅拌8 h,得到均匀的白色母液凝胶;将母液转移至水热反应釜,在200 ℃静态烘箱反应晶化48 h。晶化结束后将得到的初始产物经洗涤、干燥和焙烧后标记为SAPO-34-TM-C。
1.2 催化剂表征
采用德国布鲁克公司D8 Advance型X射线多晶衍射仪(XRD)分析分子筛物相,采用Cu/Kα为X射线源,波长为1.541 78 Å,操作电流为100 mA,操作电压为40 kV,扫描速度为10 (°)/min。采用美国安捷伦公司Varian 710-ES型电感耦合等离子体发射光谱(ICP)仪进行样品无机组分测定,光学分辨率为0.009 nm(200 nm处),分析波长为177~785 nm。为保证仪器正常运作不熄火,测试样品在HF中溶解萃取除去所有有机组分,实验测定3组平行数据取均值。采用美国FEI公司Nova Nano SEM 450型场发射扫描电镜(SEM)分析样品的表面形貌特征及晶粒尺寸大小,将干燥研磨后的分子筛镀铂60 s。在美国Micromeritics公司ASAP 2020 HD88型全自动微孔物理吸附仪上进行N2物理吸附,操作条件为:约100 mg焙烧过的分子筛先在320 ℃,真空条件下脱气24 h,然后在-196 ℃下饱和吸附氮气后脱附,得到样品的吸/脱附等温线。通过Brunauer-Emmett-Teller(BET)多点法计算样品的比表面积,t-plot法计算微孔比表面积和微孔体积,总孔体积根据相对压力(P/P0)为0.99时的吸附量估算。在美国Micromeritics公司Auto ChemⅡ型程序升温化学吸附仪上进行氨气程序升温脱附(NH3-TPD)分析分子筛样品的酸性性能,干燥后样品在550 ℃,氦气氛围下活化1 h,然后降温至100 ℃,吸附氨气30 min,氦气吹扫1 h,再以10 ℃/min的升温速率升至600 ℃。
1.3 催化性能评价
将0.1 g粒径为60~80目(0.180~0.125 mm)的催化剂装填在石英管(10 mm×45 mm)中,置于具有程序控温功能的固定床反应炉内,在N2氛围升温至550 ℃预处理5 h,然后降温至450 ℃,将质量空速(WHSV)为2 h-1的二甲醚(DME)引入反应器进行DTO反应。使用具有火焰离子化检测器(FID)和HP-PLOT色谱柱(30 m×320 μm×20 μm)的气相色谱仪在线采集气相产物进行组分分析。基于CH2计算二甲醚的转化率和产物选择性,其中甲醇被视为反应物。
2 结果与讨论
2.1 分子筛结构性能表征
用共模板方法和相转移合成,可以显著改变SAPO-34晶体的尺寸、形貌和比表面积等结构特征,在MTO(DTO)催化反应中可以提高催化剂寿命和双烯收率。为了探究在相转移合成体系中,结构导向剂用量对SAPO-34结构的调控作用,在其他原料物质的量之比不变的情况下,系统调变了原料中共模板总量及TEAOH与MOR的比例,所合成样品的XRD表征结果如图1所示。由图可知,当TEAOH与MOR物质的量之比(TEAOH/MOR比)为0.51:1.49时,有磷酸铝的杂峰出现,其他结构导向剂比例制得产物的衍射峰与CHA结构SAPO-34的衍射峰(JCPDS No. 47-0617)一致,没有出现杂峰。TEAOH/MOR比为0.34:2.66时,样品的衍射峰强度较低,可以认为TEAOH的含量较少时,制备的SAPO-34样品结晶度较低;当TEAOH/MOR比为0.51:2.49时,样品的衍射峰峰强度最高;当TEAOH/MOR比分别为1.02:0.98和1.02:1.98,总模板剂量为2和3时,两种样品结晶度均较高,且衍射峰出现明显的宽化,可认为是样品颗粒变小所致。

图1 相转移合成SAPO-34分子筛的XRD图谱Fig.1 XRD patterns for SAPO-34 zeolites synthesized by phase transfer method
采用SEM对上述探索中制得结晶度较高的纯相SAPO-34样品的形貌进行了表征,结果如图2所示。首先,3个样品均没有絮状的无定形产物,说明样品为纯相。当TEAOH/MOR比为0.51:2.49时,样品具有CHA结构的典型立方体形貌和附在立方体上生长的小颗粒及小颗粒团聚生成的不规则块状,说明TEAOH含量较低时,难以通过相转移法对SAPO-34的晶粒大小进行有效调控。TEAOH/MOR比为1.02:0.98和1.02:1.98时,样品均具有纳米级的晶粒尺寸,说明相转移合成策略在TEAOH含量较高时,对SAPO-34的微观结构有积极的影响,这与XRD的结果一致。TEAOH/MOR比为1.02:0.98时,SAPO-34晶体形貌为小的正方体颗粒,但是晶体尺寸不均一,且有团聚的大块生成;增加MOR的含量到TEAOH/MOR比为1.02:1.98时,样品具有均一的纳米小颗粒。综上,选取TEAOH/MOR比为1.02:1.98作为优化配方,所合成样品标记为SAPO-34-TM-N。

图2 相转移策略合成SAPO-34分子筛的SEM照片Fig.2 SEM images for SAPO-34 zeolites synthesized by phase transfer method
图3是普通水热体系中共模板法制备样品SAPO-34-TM-C和相转移法制备样品SAPO-34-TM-N的XRD图谱。由图可知,两种样品的衍射峰均与SAPO-34标准谱图(JCPDS No. 47-0617)的衍射峰一致,没有无定型的包峰或其他物质衍射峰出现,表明两种样品均为纯相的SAPO-34晶体。将SAPO-34-TM-C的结晶度定义为100%,通过2θ为9.5°,16.0°,20.5°,25.8°和30.5°处5个峰积分面积的比值,计算得出SAPO-34-TM-N的相对结晶度为106%,表明相转移合成的分子筛具有较高的结晶度。两种样品的元素分析结果如表1所示,通过相同原料配比所合成的SAPO-34晶体的元素组成十分接近,没有明显的差别。

图3 SAPO-34-TM-C和SAPO-34-TM-N的XRD图谱Fig.3 XRD patterns of SAPO-34-TM-C and SAPO-34-TM-N

表1 两种不同方法合成的SAPO-34分子筛的元素组成Table 1 Molar composition of the synthesized SAPO-34 from two different methods
通过SEM分析两种样品的微观结构如图4所示。两种SAPO-34的晶体中均未有层状前驱体或无定型的物相出现,说明分子筛晶化完全,这与XRD的结果相一致。SAPO-34-TM-C主要由长方体的大颗粒和附着在长方体上生长的纳米小颗粒两种形貌组成,长方体的颗粒粒径大数量少,颗粒尺寸在200~400 nm;可以清楚地看到纳米小颗粒有正方体和不规则的小块,可以认为静态的水热晶化在共模板体系中没有生成均匀的晶体颗粒。相较而言,采用相转移合成策略制备的SAPO-34-TM-N晶体形貌均一,粒径主要分布在25~70 nm,平均粒径为55 nm,具有典型的纳米堆积结构。

图4 SAPO-34-TM-C和SAPO-34-TM-N的SEM及对应的粒径分布图Fig.4 SEM images for SAPO-34-TM-C and SAPO-34-TM-N and the corresponding histogram particle size distribution
图5为水热共模板法与相转移法合成SAPO-34分子筛的N2-物理吸脱附等温线。从图中可得,在P/P0小于0.10时吸附量迅速上升,这是N2在微孔中的填充过程带来的,说明样品中存在大量微孔;在P/P0大于0.85时,曲线的上升是由于在介孔和大孔中的毛细凝结现象造成的,说明纳米晶体的堆积形成了部分介孔和大孔。SAPO-34-TM-C和SAPO-34-TM-N两个分子筛的N2-物理吸脱附等温线趋势相同,形状相近。该吸附-脱附等温线为I型和IV型的混合型,是多级孔道分子筛的典型等温线。由此可见,两类SAPO-34分子筛均具有多级孔道结构特点。

图5 SAPO-34-TM-C和SAPO-34-TM-N分子筛的N2-吸附脱附等温线Fig.5 N2 adsorption-desorption isotherms for SAPO-34-TM-C and SAPO-34-TM-N
由N2-物理吸附结果计算得到SAPO-34分子筛的孔结构数据如表2所示。从表2可以看到,SAPO-34-TM-N的比表面积从SAPO-34-TM-C的575 m2/g增加到672 m2/g,这主要来自外比表面积(从52 m2/g到116 m2/g)的增大,归因于纳米小颗粒堆积的空隙形成介孔。SAPO-34-TM-N与SAPO-34-TM-C的微孔比表面积及微孔体积基本一致,可以认为二者保持了相同的微孔结构,这与XRD测得的结晶度结果一致。总孔体积的增大主要是介孔体积的贡献,表明相转移策略可以减小分子筛粒径,提高分子筛外表面面积,并促进堆积介孔/大孔的形成。

表2 两种SAPO-34分子筛孔结构性质Table 2 Textural properties of two SAPO-34 samples
SAPO-34-TM-C和SAPO-34-TM-N的NH3-TPD曲线如图6所示。由图可知,两种SAPO-34分子筛的NH3-TPD曲线均包含两个脱附峰。193 ℃左右出现的是弱酸的信号峰,此峰来源于分子筛中对催化反应没有活性的弱酸位,443 ℃左右出现的则是具有催化活性的强酸信号峰。NH3-TPD曲线显示两种样品都具有一定的强酸位点,高温解吸曲线高点分别在443 ℃(SAPO-34-TM-C)和428 ℃(SAPO-34-TM-N);两个样品在高温段的解吸曲线峰强度存在差异,SAPO-34-N的峰强度相对较低,表明相转移策略合成的SAPO-34-TM-N分子筛的酸强度与SAPO-34-TM-C接近,但酸密度相对降低。

图6 SAPO-34-TM-C和SAPO-34-TM-N的NH3-TPD曲线Fig.6 NH3-TPD curves of SAPO-34-TM-C and SAPO-34-TM-N
2.2 催化性能评价
以二甲醚制烯烃作为探针反应,对两种不同SAPO-34分子筛的催化性能进行了评价,结果如图7和表3所示。
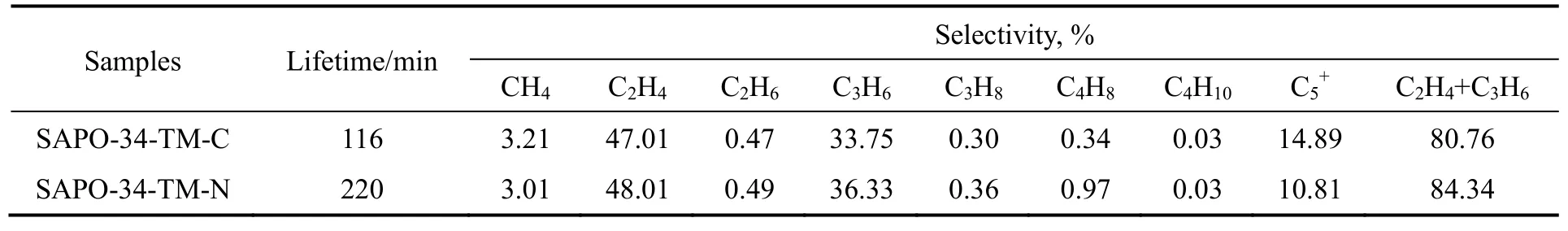
表3 SAPO-34分子筛二甲醚转化制烯烃催化数据Table 3 DTO results for SAPO-34 zeolites

图7 二甲醚转化制烯烃催化性能评价结果Fig.7 Catalytic performance of SAPO-34-TM-C and SAPO-34-TM-N in DTO.
两种催化剂都经过诱导期后提升反应活性,诱导期结束后分别进入平台期和失活期。由图7(a)和表3可以看出,SAPO-34-TM-N的催化寿命(定义为二甲醚的转化率大于99%)比SAPO-34-TM-C提高近1倍,这是由于相转移制得的催化剂能够有效地降低分子在微孔内的扩散路径,减缓孔道内“烃池”中间体向积炭前驱体的副反应,延缓失活过程,提升催化剂的稳定性,同时催化剂晶粒变小,有利于催化剂寿命延长。在双循环的反应机理中,乙烯主要通过芳烃循环产生,丙烯和丁烯在两个循环中都可生成,C5+物种只能由烯烃循环产生。在SAPO-34-TM-C上,由于催化剂晶粒较大,芳烃循环是主要的转化途径,低碳烯烃的选择性相对较高;在纳米晶粒SAPO-34-TM-N中,扩散路径的缩短使得碳链较长的烃类能够及时离开催化剂,降低继续参与甲基化-裂解反应生成低碳烯烃的可能,因此低碳烯烃的选择性下降,质量较大的产物比重增加。
从两个催化剂的催化寿命可以看出,纳米粒径和相通的晶间介孔能够改善催化剂的扩散性能,提升稳定性。与SAPO-34-TM-C相比,纳米粒径SAPO-34-TM-N对于C2H4和C3H6的总选择性(表3)也有4%左右的提高。两种催化剂上乙烯与丙烯的比例变化如图8所示。随着反应的进行,催化剂孔道内部逐渐堵塞,产物中C2H4和C3H6质量比(C2H4/C3H6比)逐渐增大。同时可以观察到在整个反应过程中,SAPO-34-TM-C的C2H4/C3H6比始终高于纳米催化剂SAPO-34-TM-N。Corma等[21,24-26]研究表明,催化剂酸性性质是决定C2H4/C3H6比的主要因素,较强的酸性性质有利于提高C2H4/C3H6的比值。因为从六甲基或四甲基苯的反应中心生成乙烯的能量较弱,所以具有较强酸性性质的SAPO-34应产生更多的乙烯,此结果和NH3-TPD的结果一致,纳米SAPO-34-TM-N酸密度较小,因此C2H4/C3H6的比例降低。

图8 DTO反应中不同SAPO-34分子筛C2H4/C3H6比随反应时间的变化Fig.8 C2H4/C3H6 ratio as a function of TOS in DTO conversion for SAPO-34-TM-C and SAPO-34-TM-N
3 结 论
四乙基氢氧化铵和吗啡啉共模板体系中,相转移策略由于成核速度快,晶体生长受到抑制,因此制得的SAPO-34-TM-N分子筛平均晶粒尺寸为55 nm,具有CHA拓扑结构典型的形貌特征,同时引入介孔孔隙,在不损失微孔性质的前提下显著增加了外比表面积和介孔体积。在二甲醚转化制烯烃探针反应中,相转移合成法得到的纳米多级结构SAPO-34-TM-N分子筛比普通水热合成的SAPO-34-TM-C分子筛具有更优的扩散和催化性能。SAPO-34-TM-N的催化寿命较SAPO-34-TM-C提升了近1倍,乙烯/丙烯的选择性也提高了约4%。
猜你喜欢
辽宁化工(2022年8期)2022-08-27
科学家(2022年4期)2022-05-10
——水热过程影响机制
化工进展(2021年11期)2021-11-30
石油炼制与化工(2021年9期)2021-09-15
建材发展导向(2021年13期)2021-07-28
石油炼制与化工(2021年3期)2021-03-23
化工学报(2020年4期)2020-05-28
东北大学学报(自然科学版)(2020年1期)2020-02-15
化工时刊(2020年11期)2020-01-12
中国惯性技术学报(2019年5期)2020-01-07
