
超高效液相色谱-串联质谱法测定牛乳中21 种β-兴奋剂
2020-07-07 09:12张立佳白艳梅刘丽君李翠枝
乳业科学与技术 2020年3期
张立佳,胡 雪,莫 楠,白艳梅,张 悦,刘丽君,李翠枝*
(内蒙古伊利实业集团股份有限公司,内蒙古 呼和浩特 010110)
β-肾上腺素受体激动剂(简称β-兴奋剂)是人工合成的苯乙醇胺类药物,主要用于治疗支气管痉挛、哮喘等疾病,由于β-兴奋剂可提高动物的饲料转化率和瘦肉率,常被非法用作动物的生长促进剂[1-3],食用含有该类物质的动物性食品会引起中毒,甚至危及生命。2002年我国农业部、卫生部、国家药品监督管理局联合发布《禁止在饲料和动物饮用水中使用的药物品种目录》,盐酸克伦特罗和莱克多巴胺等7 种“瘦肉精”为禁用药品[4]。2020年1月农业农村部公告第250号发布《食品动物中禁止使用的药品及其他化合物清单》,规定β-兴奋剂类及其盐、酯全部为禁用药品[5]。而目前现行的GB/T 22965—2008《牛奶和奶粉中12 种β-兴奋剂残留量的测定 液相色谱-串联质谱法》的检测项目仅有12 种,且前处理较复杂。因此,建立一种简单、快速、灵敏度高、能够同时检测多种β-兴奋剂的方法十分必要。
目前,测定β-兴奋剂残留量的方法有酶联免疫吸附测定法[6]、气相色谱法[7]、气相色谱-串联质谱法[8-9]、生物芯片技术[10]、高效液相色谱-串联质谱法(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)[11-16]和超高效液相色谱-串联质谱法(ultra-high performance liquid chromatographytandem mass spectrometry,UPLC-MS/MS)[17]等。UPLC-MS/MS具有高效、特异性强、灵敏度高、多种化合物同时检测等优势,是β-兴奋剂最主要的检测方法。本研究通过对前处理方法和仪器条件的优化,确立UPLC-MS/MS同时检测牛乳中21 种β-兴奋剂的方法。
1 材料与方法
1.1 材料与试剂
牛乳(500 g) 市售。
β-葡萄糖醛酸酶/芳基硫酸酯酶、乙酸铵(色谱纯)上海安谱实验科技股份有限公司;乙腈(色谱纯) 德国Meker公司;甲醇(色谱纯) 美国Fisher公司;甲酸(色谱纯) 天津市沃尔孚科技发展有限公司;21 种标准品(马布特罗、妥布特罗、氯丙那林、西布特罗、溴布特罗、喷布特罗、克伦普罗、特布他林、非诺特罗、齐帕特罗、班布特罗、苯乙醇胺A、克仑潘特、羟甲基氨克仑特罗、苯氧丙酚胺、沙丁胺醇、利托君、莱克多巴胺、克伦特罗、西马特罗、沙美特罗)德国Dr.Ehrenstorfer公司;沙丁胺醇-d3(salbutamol-d3,SOL-d3)、莱克多巴胺-d3(ractopamine-d3,RNE-d3)、克伦特罗-d9(clenbutrol-d9,CIB-d9) 美国Waters公司;实验用水为超纯水。
1.2 仪器与设备
U P L C-M S/M S 仪(配备电喷雾离子源)、Oasis MCX固相萃取柱(60 mg/3 mL)、BEH C18柱(5 0 m m×2.1 m m,1.7 μ m)、B E H C18柱(100 mm×2.1 mm,1.7 μm) 美国Waters公司;3K30低温高速离心机 德国Sigma公司;KS 501涡旋混合振荡器 德国IKA公司;10430 Harris Oaks氮吹仪 瑞典Biotage公司;固相萃取装置 美国Supelco公司;FE20 pH测定仪 梅特勒-托利多仪器(上海)有限公司。
1.3 方法
1.3.1 样品前处理
1.3.1.1 提取
称取5 g牛乳样品于50 mL离心管中,加入100 μL 3 种混合同位素内标(质量浓度均为10 ng/mL)静置10 min,加入15 mL 0.2 mol/L乙酸钠-醋酸缓冲液(pH 5.2±0.2)和100 μLβ-葡萄糖醛苷酶/芳基硫酸酯酶溶液(10 000 U/mg),在(37.0±0.5) ℃水浴中酶解12 h后取出恢复至室温,用5 mol/L盐酸溶液调节酶解后溶液pH值至2.0±0.1,10 000 r/min离心10 min,分离上层溶液(如果液面有脂肪析出,可以用棉花过滤),转移到50 mL离心管,12 000 r/min离心10 min,分离上清液,待净化。
1.3.1.2 净化
依次用3 mL甲醇、3 mL水、3 mL 40 mmol/L盐酸溶液活化平衡固相萃取柱,将上清液全部转移至固相萃取小柱净化,然后依次用3 mL水、3 mL甲醇淋洗除去杂质,用真空泵抽干柱内液体,至少30 min,加入10 mL 5%氨化乙酸乙酯(浓氨水、乙酸乙酯体积比5∶95,下同)洗脱(充分混匀后,静置到溶液透明至底层有分层现象再使用,取上层有机层,水层不能作为洗脱液使用),洗脱液收集于12 mL氮吹管中,在40 ℃水浴中氮吹至近干,加入1 mL 0.1%甲酸-10%甲醇水溶液混匀1 min,过0.22 μm有机滤膜后,待UPLC-MS/MS分析。
1.3.1.3 标准曲线的绘制
用甲醇溶解β-兴奋剂标准品,配成质量浓度为100 mg/L的单标储备液,于―18 ℃条件下保存备用。使用时根据需要配成质量浓度100 ng/mL的混合标准中间液,将混合标准中间液用1 mL 0.1%甲酸-10%甲醇水溶液逐级稀释得到质量浓度分别为0.2、0.5、1.0、2.0、5.0、10.0 ng/mL的混合标准工作液,同时分别加入3 种混合内标中间液(质量浓度均为10 ng/mL)。质量浓度由低到高进行检测,以定量离子响应值-质量浓度作图,得到标准曲线回归方程。
1.3.2 色谱条件
色谱柱:Acquity UPLC BEH C18柱(50 mm×2.1 mm,1.7 μm);柱温35 ℃;进样量10 μL;流动相A为体积分数0.1%甲酸水溶液,流动相B为甲醇;流速0.3 mL/min;线性梯度洗脱条件如表1所示。
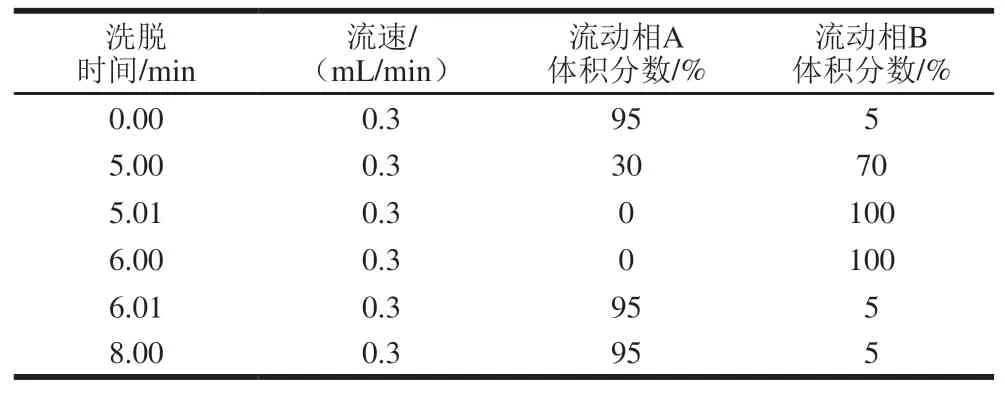
表 1 梯度洗脱条件Table 1 Procedure of gradient elution
1.3.3 质谱条件
电离模式:电喷雾离子源正离子模式,毛细管电压3.5 kV,离子源温度150 ℃,脱溶剂温度500 ℃,脱溶剂气流量650 L/h,碰撞室气流量0.18 mL/min,多反应监测模式检测,其他质谱条件如表2所示。
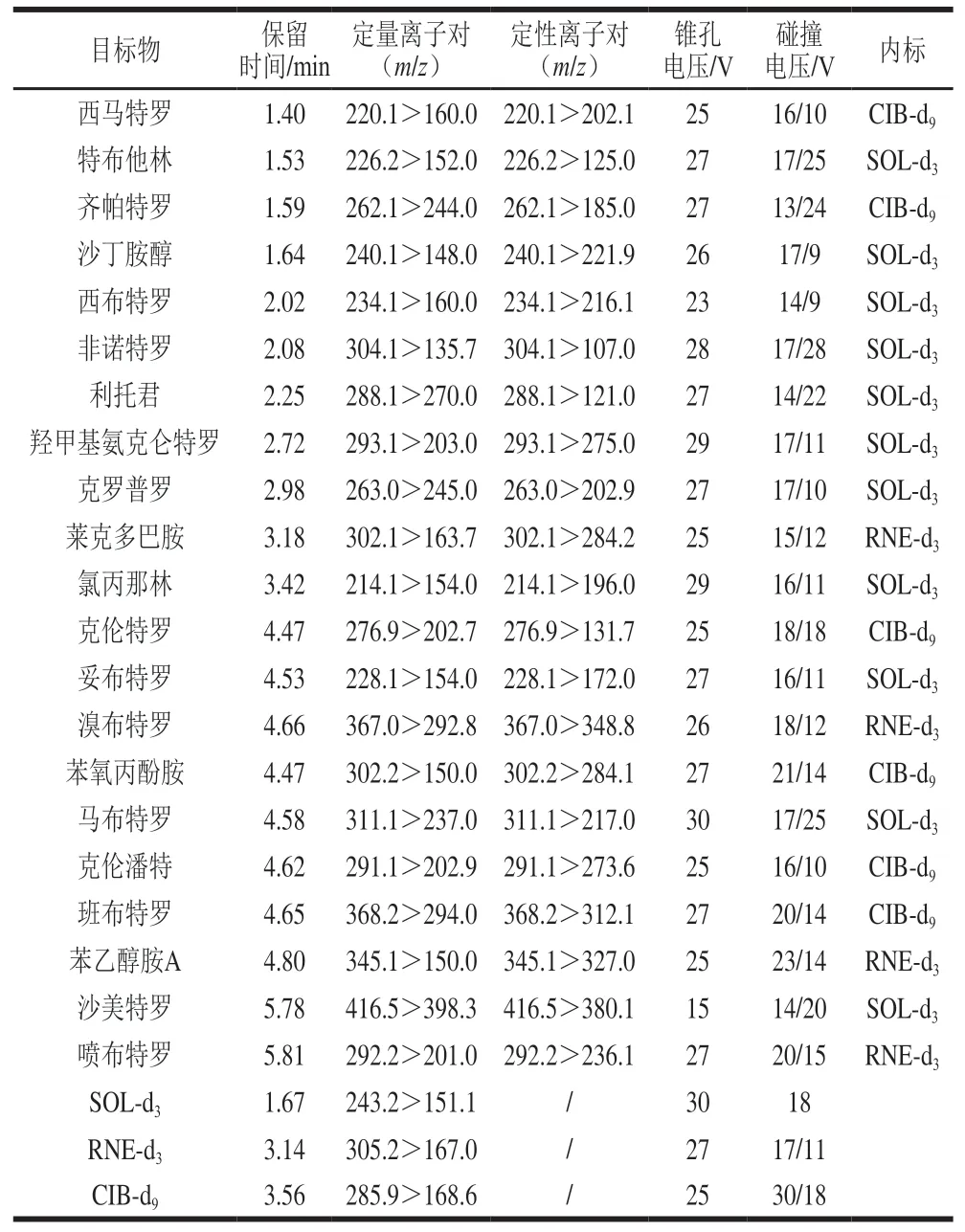
表 2 21 种β-兴奋剂及同位素内标质谱条件Table 2 Mass spectrometric conditions for 21β-agonists and isotopic internal standards
1.3.4 结果计算
试样中β-兴奋剂的含量按下式计算:
式中:X为试样中β-兴奋剂残留量/(μg/kg);ρ为上机溶液中β-兴奋剂残留量/(ng/mL);V为样液最终定容体积/mL;n为稀释倍数;m为试样质量/g。
2 结果与分析
2.1 前处理方法的选择
2.1.1 预处理液的选择
牛乳样品酶解后,酶解液中分别加入一定体积的体积分数0.2%乙酸水溶液、0.2 mol/L高氯酸水溶液和5 mol/L盐酸溶液,进行蛋白沉淀和提取目标物。结果表明:加入体积分数0.2%乙酸水溶液时,克伦特罗受基质干扰较大,目标峰与基质干扰峰无法分离(图1A);加入0.2 mol/L高氯酸水溶液时,克伦特罗目标峰受基质干扰,未完全分离(图1B);加入5 mol/L盐酸溶液(pH 2.0)时,克伦特罗基质干扰较小,目标峰响应明显增强,检出限满足方法要求(图1C),同时,其余20 种化合物色谱峰均得到改善,满足方法定量限。继续考察用5 mol/L盐酸溶液调节酶解液pH值至1.0、2.0、3.0、4.0,结果表明:当调节pH值至3.0和4.0时,离心后效果均较差,无法沉淀牛乳中的蛋白;当调节pH值至1.0和2.0时,21 种化合物的检出限均能达标,且21 种化合物的回收率无明显差异。为了减少酸的用量,最终选择5 mol/L盐酸溶液(pH 2.0)作为预处理液。
2.1.2 净化方法的选择
根据化合物性质及相关标准(GB/T 22965—2008《牛奶和奶粉中12 种β-兴奋剂残留量的测定 液相色谱-串联质谱法》[18]、GB/T 21313—2007《动物源性食品中β-兴奋剂残留检测方法 液相色谱-质谱/质谱法》[19]),选择离子交换柱(Oasis MCX)进行净化,需要选择偏碱性的氨化溶剂作为洗脱液,对比5%氨化乙酸乙酯和5%氨化甲醇(浓氨水、甲醇体积比5∶95,下同)作为洗脱液的洗脱效果。结果表明:选择5%氨化甲醇作为洗脱液时,化合物受基质影响较明显,尤其是沙丁胺醇和克伦特罗有较强的干扰峰,检出限无法满足方法要求;选择5%氨化乙酸乙酯作为洗脱液时,极性较强的化合物保留在色谱柱上,不会被洗脱下来,克伦特罗和沙丁胺醇受基质干扰的影响均得到改善,且21 种化合物的检出限均满足方法要求。因此,选择5%氨化乙酸乙酯作为洗脱液。
对5%氨化乙酸乙酯洗脱液的体积进行优化,分别考察添加6、8、10、12 mL的洗脱效果,结果表明:洗脱液体积小于10 mL,部分化合物洗脱不完全,回收率低于60%;洗脱液体积为10 mL和12 mL时,21 种化合物回收率均大于60%,由于12 mL洗脱体积过大不利于前处理,因此选择10 mL作为洗脱液体积。
此外,考察复溶液对检测结果的影响,当甲醇水溶液体积分数大于20%,西马特罗、特布他林、沙丁胺醇等出峰较早的化合物具有较强的溶剂效应,峰型较差,有严重的拖尾现象;加入适量甲酸明显提高了仪器灵敏度,最终选择0.1%甲酸-10%甲醇水溶液作为复溶液。
2.1.3 定量方法的选择
质谱检测较大优势是用目标化合物的同位素作为内标,可以校准前处理损失、消除基质效应及校准仪器系统误差,但是同位素内标成本较高,有些同位素内标合成较困难,无法商品化,多组分同时检测时,选择有代表性的几个化合物同位素内标作为共用的同位素内标是一种较为经济、简单的方法。通过对仪器条件的优化,最终选择SOL-d3、RNE-d3、CIB-d93 个同位素内标作为共用内标。
2.2 质谱条件优化
采用直接进样方式,0.01 mg/L单标进样,进样量10 μL,采用正负离子扫描模式对相应模式的母离子进行扫描,待测物响应准分子离子峰在正离子模式下获得,母离子均为[M+H]+,以准分子离子为母离子进行二级质谱扫描,得到各个化合物响应最佳的碎片离子。选择母离子及对应产生的2 个响应较强的子离子作为定性定量依据,完全满足欧盟2002/657/EC指令[20]。
2.3 色谱条件优化
主要考察Waters BEH C18柱(50 mm×2.1 mm,1.7 μm)和Waters BEH C18柱(100 mm×2.1 mm,1.7 μm)对待测样品的分析效果,结果表明:2 种色谱柱分离效果没有明显差异,Waters BEH C18柱(50 mm×2.1 mm,1.7 μm)系统背压相对较低,仪器的平衡时间具有一定优势,因此选择Waters BEH C18柱(50 mm×2.1 mm,1.7 μm)作为色谱柱。通过对洗脱梯度的多次优化,改善了克伦特罗色谱峰受基质干扰的问题,最终实现了21 种β-兴奋剂的有效分离,均得到良好的峰形,同时,缩短了分析时间,一次分析仅需8 min。
2.4 标准曲线和定量限
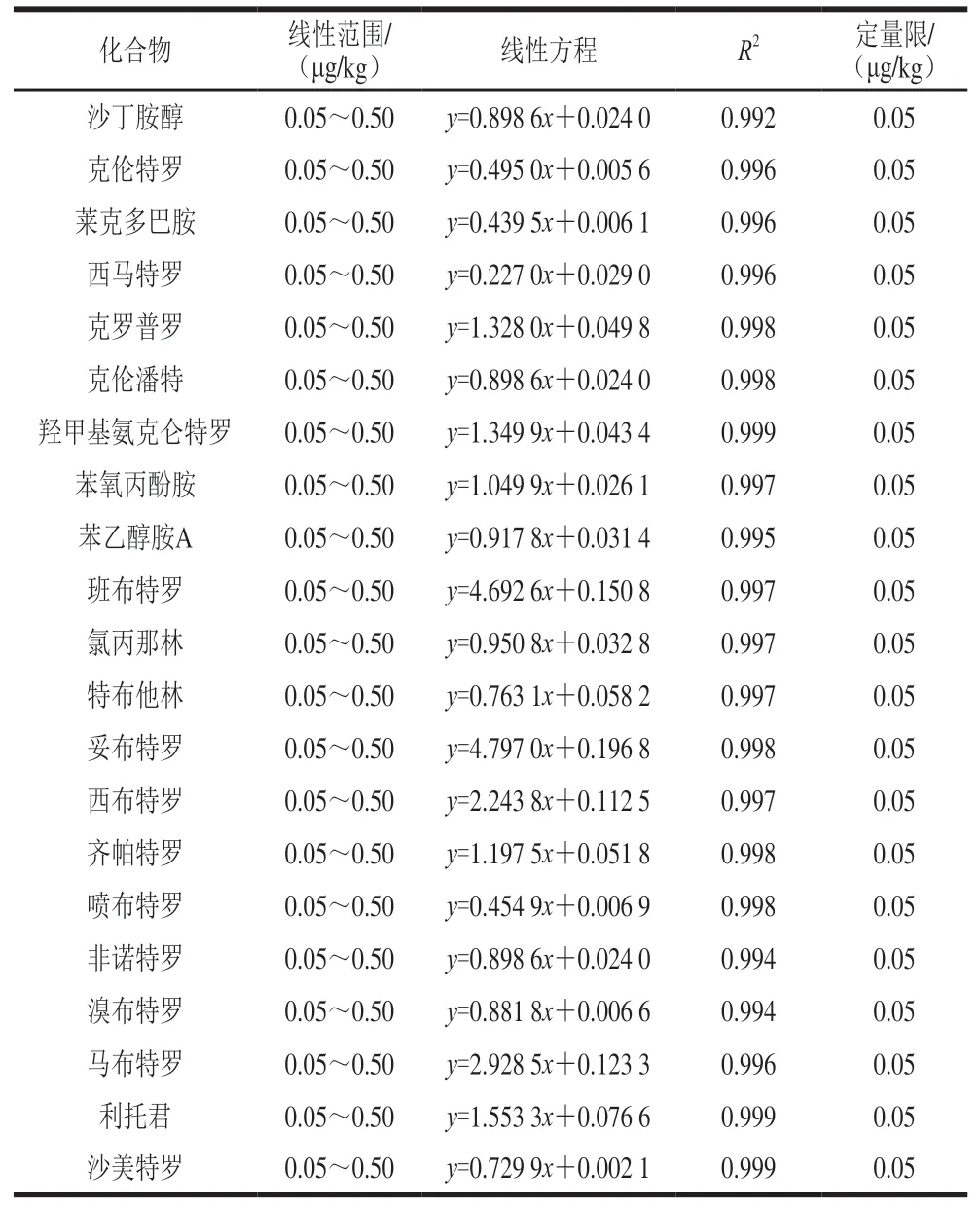
表 3 21 种β-兴奋剂的线性范围、线性方程及定量限Table 3 Linear ranges, linear equations, and limits of detection of 21 β-agonists
由表3可知,21 种β-兴奋剂添加量为0.05~0.50 μg/kg时,线性关系均良好,标准曲线相关系数(R2)均大于0.99,在牛乳中的定量限均为0.05 μg/kg,可以满足定量分析的需要。
2.5 方法回收率和精密度
测定定量限、2 倍定量限、10 倍定量限3 个不同添加水平的加标回收率。在阴性样品中分别添加0.05、0.10、0.50 μg/kg β-兴奋剂,各添加水平均做6 次平行实验,计算回收率和精密度。
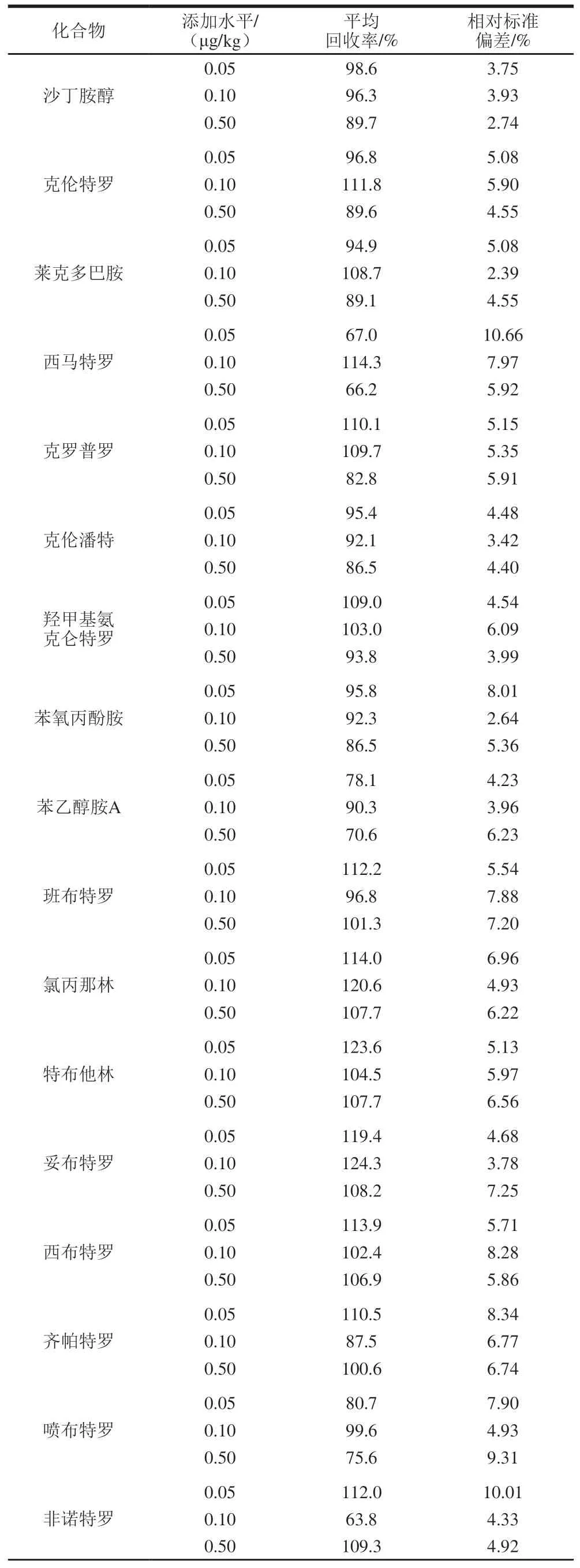
表 4 方法回收率和相对标准偏差Table 4 Recovery and relative standard deviation for spiked samples
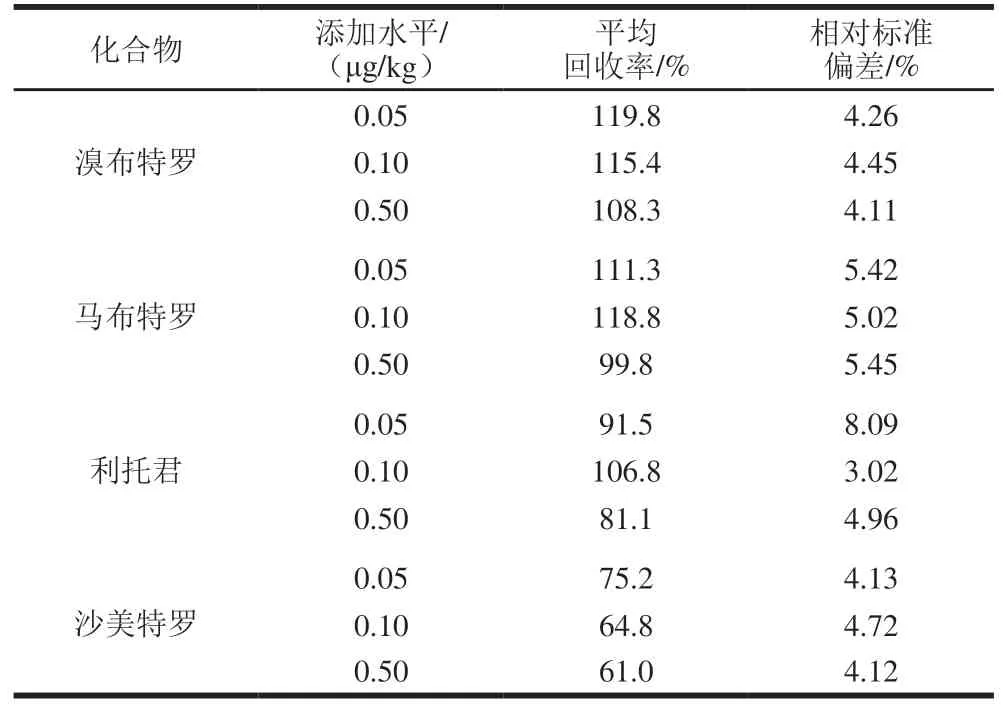
续表4
由表4可知,平均回收率为61.0%~124.3%,相对标准偏差为2.39%~10.66%,满足方法要求。
2.6 实际样品测定
利用本研究建立的前处理和UPLC-MS/MS方法对市场上销售的20 批次成品牛乳进行检测,21 种β-兴奋剂均未检出。
3 结 论
采用UPLC-MS/MS法同时测定牛乳中21 种β-兴奋剂的含量,牛乳中21 种β-兴奋剂的定量限均为0.05 μg/kg,在0.05、0.10、0.50 μg/kg 3 个不同添加水平下,基质平均回收率为61.0%~124.3%,相对标准偏差为2.39%~10.66%。本方法简便、快速、回收率高、重复性好,可以准确测定牛乳中β-兴奋剂的含量。
猜你喜欢
体育科技文献通报(2022年3期)2022-05-23
口腔护理用品工业(2021年4期)2021-11-02
食品安全导刊(2021年21期)2021-08-30
成都体育学院学报(2021年1期)2021-07-16
食品界(2019年2期)2019-03-10
中国科技纵横(2019年23期)2019-02-14
海峡科技与产业(2017年1期)2017-03-04
食品工业科技(2014年5期)2014-03-11
中国信息化·学术版(2013年3期)2013-06-25
中国体育(2004年3期)2004-11-11
