
MgO改性无粘结剂ZSM-5分子筛制备及催化苯与乙烯烷基化反应性能
2020-07-05 01:12马翀玮沈震浩蔡焕焕王达锐孙洪敏杨为民朱志荣
化学反应工程与工艺 2020年4期
马翀玮,沈震浩,蔡焕焕,王达锐,孙洪敏,杨为民,朱志荣
1.华东理工大学化工学院,上海 200237;2.绿色化工与工业催化国家重点实验室,中国石油化工股份有限公司上海石油化工研究院,上海 201208;3.同济大学,化学科学与工程学院,上海 200092
对二乙苯是一种高附加值有机化工原料,主要用作对二甲苯生产过程中的解吸剂以及聚乙烯的改进剂,也是生产高端共聚物如离子交换树脂、交联剂等的重要单体[1]。目前工业生产对二乙苯的方法主要为直接合成法和吸附分离法[2],直接合成法又分为乙苯歧化法和乙苯烷基化法。
生产对二乙苯的原料乙苯是重要的大宗化工中间体,主要用于生产苯乙烯[3]。工业上乙苯的制备通常采用苯与乙烯在MFI型分子筛(ZSM-5)上的气相烷基化或在MWW型等分子筛上的液相烷基化方法[4-5],而过程中的二次烷基化反应会不可避免地生成二乙苯,尽管它们可通过与苯的烷基转移来回收乙苯,但也增加了一定的生产成本。若通过苯与乙烯烷基化法,在生产乙苯的同时联产对二乙苯,则又受到催化剂性能的制约,经济性较差。
二乙苯有三种主要异构体,即邻二乙苯(分子直径d为0.78 nm,)、间二乙苯(d为0.72 nm)和对二乙苯(d为0.56 nm)。ZSM-5分子筛为具有两种相互交叉的十元环孔道体系,平行于b轴的截面呈椭圆形的直孔道(孔口截面约0.54 nm×0.56 nm),平行于a轴和c轴的截面近似为圆形的正弦孔道(孔口截面约0.54 nm×0.51 nm),根据分子筛择形催化过渡态限制的原理,ZSM-5可以实现烷基化或歧化反应的对位择形性[6-7]。但由于二乙苯会在催化剂外表面酸性位上发生非择形的异构化反应,生成间二乙苯和邻二乙苯,最终导致对位选择性下降。
化学气相沉积法(CVD)[8-10]、化学液相沉积法(CLD)[11-14]、预积炭[15]和金属化合物浸渍[16-20]等方法常被用来钝化ZSM-5外表面的酸性位,提高对位择形选择性。气、液相沉积法虽然能很大程度地提高对位选择性,但会造成分子筛孔口缩小,大幅度缩短催化剂使用寿命。预积炭方法在每次催化剂再生后需重复积炭操作且积炭效果难以控制。而金属化合物浸渍可以覆盖分子筛外表面活性位,减弱表面酸性,缩小孔口,从而提高对位选择性,某些金属化合物还可以对催化剂孔道内的强酸位进行修饰,减弱酸性,减少副反应的发生。苯与乙烯气相烷基化反应中,副反应较为复杂,易生成不易分离去除的关键杂质二甲苯,金属化合物浸渍可能会对该反应有好的改性效果。
前期工作中已成功合成了无粘结剂核壳ZSM-5分子筛催化剂,与含有粘结剂的样品相比,无粘结剂核壳ZSM-5分子筛催化剂扩散效果好,酸性有效分散,副反应减少,稳定性提高[21]。因此本工作在此工作基础上,进一步研究MgO浸渍改性对核壳无粘结剂ZSM-5分子筛催化剂物化性质的影响,考察其在苯与乙烯烷基化合成乙苯并联产对二乙苯反应中的催化性能,以揭示MgO改性对反应活性、择形选择性、稳定性等性能的影响规律。
1 实验部分
1.1 催化材料制备
将Na型ZSM-5分子筛与粘结剂(质量分数40%的硅溶胶)均匀捏合,挤条成型得到直径为2.0 mm圆柱体条形颗粒,然后将颗粒在150 ℃下烘干6 h,并在550 ℃下焙烧5 h,得粘结剂SiO2质量百分比为35%的催化剂。制得的样品在1.0 mol/L的NH4Cl溶液中,90 ℃离子交换1 h,并重复交换操作三次,然后在150 ℃下烘干6 h,并于550 ℃下焙烧5 h,得到H型ZSM-5催化剂样品,标记为S0。
将样品S0通过浸渍法进行MgO改性。将样品S0在质量分数为17.7%的乙酸镁水溶液中浸渍12 h,过滤后在40 ℃下干燥8 h,并在100 ℃下连续干燥12 h,然后在550 ℃下焙烧6 h,得到MgO改性的ZSM-5催化剂,标记为S0-MgO。
根据气固相二次晶化方法[21],以样品S0为前驱体,乙胺(质量含量65%)为结构导向剂,采用蒸汽相晶化方式使粘接剂转化结晶,合成了无粘结剂核壳ZSM-5催化剂,标记为S1,并采用与S0-MgO相同的浸渍改性方法制备MgO改性的S1-MgO样品。
1.2 分子筛表征
样品的X射线衍射(XRD)分析在Bruker D8 Advanced射线衍射仪进行。X射线源为0.154 18 nm Cu/Kα,管电流为40 mA,管电压40 kV,扫描2θ为2°~45°,步长0.05°,扫描速度4 (°)/min。扫描电子显微镜(SEM)在FEI Nova NanoSEM 450型电子显微镜中进行。N2吸附-脱附等温线在Micromeritics ASAP 2010物理吸附仪中进行,在300 ℃下抽真空,采用Brunauer-Emmett-Teller(BET)公式和Barrett-Joyner-Halenda(BJH)模型计算样品的比表面积、孔径分布和孔容,t-plot曲线法计算微孔体积和微孔比表面积。样品的体相硅铝物质的量比和Mg含量采用化学分析结合电感耦合等离子体法(ICP,Varian 725-ES)进行测定。样品的酸性表征在天津鹏翔PX200系列多用吸附仪上进行氨气程序升温脱附实验,脱附温度为100~600 ℃,升温速率为10 ℃/min。催化剂酸性在Nicolet NEXUS470 FTIR 红外光谱仪上采用吡啶吸附-脱附傅立叶变换红外测定。压片制得质量15 mg催化剂薄片,在红外池中400 ℃脱附4 h,依次记录400,300和200 ℃时的本体背景,再吸附吡啶,低压抽真空30 min,而后升温至300 ℃和400 ℃,分别记录脱附本体谱图。样品的探针反应[异丙苯(IPB)和1,3,5-三异丙苯(TIPB)裂解反应]实验在固定床微型反应器中进行。催化剂装填量为50 mg,液态异丙苯和液态1,3,5-三异丙苯的进料量均为1 μL,常压,气化温度为150 ℃,反应温度为350 ℃。采用配备氢离子火焰检测器(FID)的气相(Agilent GC 7890)色谱分析产物,色谱柱为HP-FFAP,氩气为载气,流量为20 mL/min。根据产物的峰面积计算原料的转化率。
1.3 分子筛催化性能评价
苯与乙烯气相烷基化反应在固定床不锈钢反应器(内径为13 mm)中进行。催化剂用量为0.6 g,苯与乙烯物质的量之比为7:1,反应温度为380 ℃,物料总气空速为5 500 h-1。将催化剂破碎后过筛,取12~20目(0.9~1.7 mm)的颗粒装填,以氮气为载气,在380 ℃下预处理2 h,通入苯进行填充并升压至1.5 MPa,恒温恒压0.5 h后,通入乙烯开始烷基化反应。产物采用Agilent GC 7890色谱仪在线分析,氢离子火焰检测器(FID),采用配有Agilent Dean Switch 技术的双色谱柱分析方法,对轻质非芳烃和芳烃分别进行分离。轻质非芳烃采用HP-Plot-Q毛细色谱柱进行分离,使乙烯/乙烷、丙烯/丙烷等难分离的物质有效分离;芳烃则采用HP-FFAP毛细色谱柱进行分离;所有有机产物的峰面积归一化处理。分别以CEthylene,SEthyl,SEB和SXylene来表示苯与乙烯气相烷基化反应中的乙烯转化率、乙基选择性[乙苯(EB)和二乙苯(DEB)选择性]、乙苯选择性和二甲苯(Xylene)选择性。选择性(Si)定义为产物中物质i的质量百分比Xi与反应消耗的苯的质量百分比(XBenzene,reacted)的比值;择形选择性(Sj-DEB)定义为间位、对位和邻位(m,p,o)二乙苯质量百分比(Xj-DEB)与总二乙苯质量百分比(XDEB)的比值。X为某一物质在反应流出物中以色谱归一化法确定的质量百分比。
2 结果与讨论
2.1 催化材料物性表征
制得的催化材料样品的氮气物理吸附结果见图1和表1。结果表明,无粘结剂核壳ZSM-5催化剂S1和其改性后的S1-MgO均表现出I型吸附等温线特性;MgO改性后的样品相较于S1比表面积从361 m2/g下降了5.8%,总孔容从0.218 cm3/g下降了6.0%,微孔孔容从0.134 cm3/g下降了4.5%。S0和其MgO改性后的样品S0-MgO表现出I型和IV型混合体的性质,表明其同时存在ZSM-5分子筛微孔结构和无定形二氧化硅所提供的介孔。MgO改性后的样品相较于S0比表面积从323 m2/g下降了8.7%,总孔容从0.296 cm3/g下降了7.1%,微孔孔容从0.111 cm3/g下降了6.3%。S0和S1在MgO改性后微孔比表面积、外表面比表面积和微孔孔容等均出现了下降的现象,表明MgO不仅覆盖了ZSM-5外表面,还进入了ZSM-5微孔中。其中S1样品的微孔受到影响小于S0,这是由于S1不含粘结剂,具有较为完整的微孔结构,而S0中的大量粘结剂对晶粒间微孔的贯通性造成了一定的影响,因此S1-MgO的微孔比表面积、微孔孔容远高于S0-MgO。元素分析(ICP)结果表明,四个样品的硅铝比基本相同,两个MgO改性后样品的镁负载量接近,分别为1.77%和1.75%。

图1 样品的氮气物理吸附-脱附曲线Fig.1 N2-adsorption and desorption isotherms of the samples
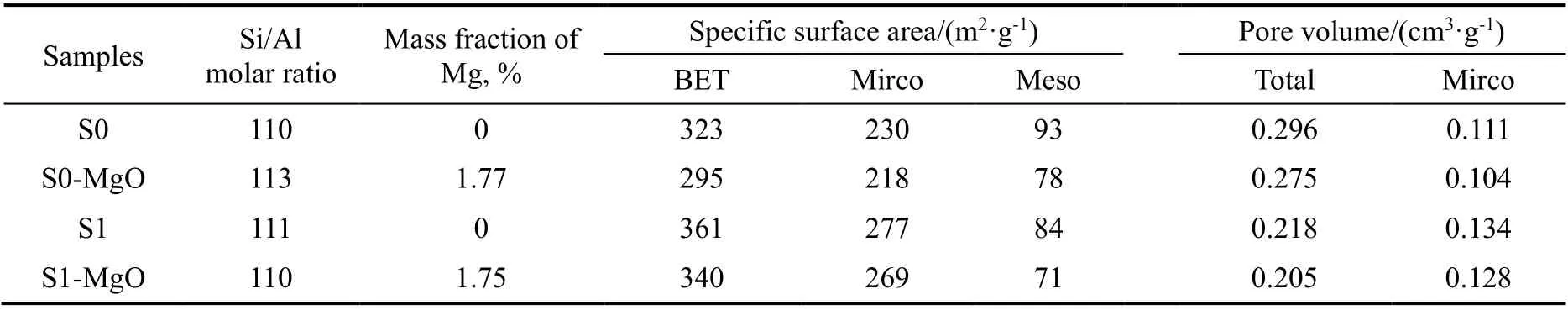
表1 样品的物理化学性质表征结果Table 1 Physicochemical properties of the samples
图2是催化剂样品的SEM照片。结果显示,S0外表面覆盖有明显的粘结剂颗粒,而S1则为ZSM-5分子筛的六方体结构,粒子大小和形状较为均一,平均尺寸约为500 nm,S0中无定型的二氧化硅粘结剂通过气固相晶化为MFI结构的ZSM-5分子筛外壳[21]。S0和S1经MgO改性后,晶体形貌和尺寸均无明显变化。

图2 样品的SEM照片Fig.2 SEM photos of the samples
图3为不同组成结构催化剂样品的XRD谱图。结果表明,S0和S1样品在2θ为7°~10°时存在两个衍射峰,在2θ为22.5°~25°时存在三个衍射峰,对应了MFI结构分子筛特征的晶相结构。由于S0样品中含有无定型二氧化硅,所以相对结晶度明显低于S1样品。经过MgO改性后的S0和S1样品,ZSM-5的特征峰强度稍有降低,但是晶相结构仍完整。另外,在42.5°左右并没有出现MgO的特征峰。这与Wang等[22]报道的MgO分散量只有高于单层分散阈值(0.06 g-MgO/g-ZSM-5)时,XRD衍射图才会出现MgO晶相的衍射峰一致。因此,上述这些结果表明通过浸渍,MgO高度分散在ZSM-5外表面和孔道中。

图3 样品的XRD图谱Fig.3 XRD patterns of the samples
2.2 酸性表征
图4为催化剂样品的氨气程序升温脱附(NH3-TPD)图谱。结果表明,所有的样品都展现出的典型双峰特性。S0和S1分别在160 ℃(低温峰)和330 ℃(高温峰)有两个特征峰,分别代表吸附在弱酸位上氨的脱附和吸附在中-强酸位上氨的脱附。S0和S1用MgO改性后,中-强酸中心峰面积大幅下降,表明MgO分散在ZSM-5分子筛的内外表面,覆盖了ZSM-5的部分中-强酸性位。而弱酸中心峰面积有所上升,并朝着高温区移动,这主要是由于镁改性的样品中Mg2+取代了分子筛中硅铝桥式羟基中的H+,形成了Mg(OH)+物种,导致L酸性位点的增加[23]。S1-MgO相较于S0-MgO,弱酸中心峰面积基本没有变化,而中-强酸中心峰面积下降,这说明了MgO更好地与无粘结剂ZSM-5分子筛的酸性位发生了作用,中-强酸中心数量减少,并且整体酸强度有所减弱。

图4 样品的NH3-TPD图谱Fig.4 NH3-TPD profiles of the samples
四个样品300 ℃时吡啶吸附红外图谱如图5所示。图5中1 550 cm-1附近的特征峰代表了B酸中心,在1 450 cm-1处的特征峰代表了L酸中心,其中S0和S1的B酸和L酸酸量相近。而S0和S1利用MgO改性后,B酸数量减少,L酸数量明显增加。这说明了MgO覆盖了ZSM-5的部分中-强酸酸性位,并且Mg2+取代了分子筛中硅铝桥式羟基中的H+,形成L酸。样品不同脱附温度的L酸和B酸酸量如表2所示。由表2可知,S0和S1经MgO改性后,L酸位吸附的吡啶更难脱附,说明新形成的L酸位具有较强的酸强度,这可以解释经MgO改性S0和S1样品NH3-TPD低温脱附峰向高温区展宽的现象;B酸位的酸强度虽然并没有减弱,但是酸量明显减少,使催化剂整体酸性降低。

图5 样品的吡啶吸附红外图谱Fig.5 Infrared spectrum of pyridine adsorption of the samples

表2 样品不同脱附温度的L酸酸量和B酸酸量Table 2 The amount of L acid and B acid of samples at different desorption temperatures
四个样品的探针分子裂解反应结果见表3。结果显示,S0和S1催化异丙苯的转化率接近,表示两者具有相近的体相酸性;而S0催化1,3,5-三异丙苯的转化率略微低于S1,这是由于1,3,5-三异丙苯的裂解反应主要发生在ZSM-5外表面的酸性位上[24],骨架Al原子在气固相晶化过程中会向外层迁移[21]。而经过MgO改性后,S0-MgO和S1-MgO催化的异丙苯转化率接近,但是后者催化1,3,5-三异丙苯的转化率明显低于前者,表明S1-MgO外表面的酸性中心覆盖程度高于S0-MgO。
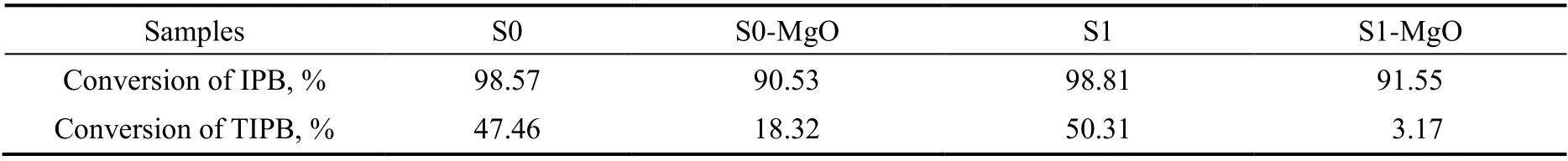
表3 样品的探针反应性能结果Table 3 Catalytic activities of samples for cracking of IPB and TIPB
2.3 苯与乙烯择形烷基化催化性能
在反应温度为380 ℃,反应压力为1.5 MPa,苯与乙烯物质的量之比为7:1,总体积空速为5 500 h-1的条件下,不同ZSM-5分子筛样品催化苯与乙烯气相烷基化反应性能如表4所示。结果表明,MgO改性后,乙烯转化率略有下降,但乙基选择性和乙苯选择性都有所上升,关键控制副产杂质二甲苯含量(选择性)下降,这主要是由于MgO改性使ZSM-5本体酸量减少,整体酸强度减弱,导致副反应发生几率降低,减少了二甲苯和其它杂质的生成。在二乙苯择形选择性方面,由于催化剂外表面酸性的钝化,S1-MgO的对位选择性明显高于S1,由40.89%提高至95.49%。二次晶化后催化剂样品中分子筛外表面更为规整,孔道扩散性较好,浸渍过程中MgO也能够更好地分散在催化剂中分子筛外表面的酸性位上,因此S1-MgO上的对二乙苯择形选择性也明显高于S0-MgO。此外,由于S1-MgO不含粘结剂,拥有更多分子筛特征微孔与晶间介孔及更好的扩散性能,使生成乙苯能够快速地从催化剂中脱附,减少了副反应发生的几率,因此其乙烯转化率、乙基选择性和乙苯选择性也均优于S0-MgO,副产二甲苯选择性也明显减少。

表4 催化剂样品在苯与乙烯气相烷基化反应中的催化性能Table 4 Catalytic performance of the samples on benzene alkylation with ethylene
在反应温度为380 ℃,反应压力为1.5 MPa,苯与乙烯物质的量之比为7:1,总体积空速为5 500 h-1的条件下,催化剂的稳定性实验结果见图6。图6(a)结果表明,三个催化剂样品得到的初始乙烯转化率都在99%以上;随着连续运行反应,S1的乙烯转化率基本不变,而MgO改性催化剂的稳定性变差,但S1-MgO的稳定性优于S0-MgO,在180 h时还能保持90%以上的乙烯转化率,而S0-MgO在60 h时已出现明显衰减,在120 h时已完全失活。图6(b)的对位择形选择性结果表明,三个样品催化的苯与乙烯气相烷基化反应中,对二乙苯择形选择性都随着连续运行反应时间增长而略有增大,这是因为随着反应的进行,分子筛催化剂孔道内逐渐积炭,导致催化剂失活,增加了二乙苯的对位选择性;但是S0-MgO的对二乙苯选择性上升较大(75%~82%),表明积炭速率较快,所以容易导致催化剂失活,S1与S1-MgO的对二乙苯择形选择性上升速率较慢,反应180 h内的变化分别为38%~41%和93%~97%,失活速率较慢,与图6(a)一致。由于具有较高的对二乙苯择形选择性和较好的稳定性,S1-MgO可在苯与乙烯烷基化制乙苯联产对二乙苯反应中长周期运行。

图6 不同催化剂样品催化苯与乙烯择形烷基化反应稳定性Fig.6 Stability of shape selective alkylation of benzene and ethylene over different catalysts
3 结 论
采用气固相晶化法使成型ZSM-5催化剂中粘接剂结晶转化,合成了无粘结剂ZSM-5分子筛催化剂条形颗粒,并采用浸渍法对其进行改性,制备了MgO改性无粘结剂ZSM-5分子筛催化剂。
MgO改性不仅钝化了ZSM-5分子筛外表面酸性,还进入孔道而使分子筛酸量减少,使得整体酸强度减弱;相较于含粘结剂的分子筛,MgO改性后的无粘结剂ZSM-5分子筛具有更好的MgO改性效果。在苯与乙烯烷基化制乙苯并联产对二乙苯反应中,MgO改性的无粘结剂ZSM-5分子筛具有较好的择形催化性能。MgO改性后,乙烯转化率略有下降,但乙基选择性和乙苯选择性都有所提高,关键控制副产杂质二甲苯选择性下降。相较于含粘结剂的样品,MgO更好地覆盖了无粘结剂ZSM-5分子筛外表面的酸性位,具有更好的对位择形选择性,在反应温度为380 ℃,压力为1.5 MPa,苯与乙烯物质的量之比为7:1,总气时空速为5 500 h-1的条件下,乙烯转化率大于99%,对二乙苯择形选择性达到93%~97%,反应稳定性明显优于含粘结剂的样品。
猜你喜欢
轻工标准与质量(2022年5期)2022-10-31
炼油与化工(2022年4期)2022-10-10
化工时刊(2022年7期)2022-09-01
辽宁化工(2022年8期)2022-08-27
中国钢铁业(2022年3期)2022-07-06
炼油技术与工程(2022年2期)2022-04-14
黑龙江交通科技(2021年10期)2021-11-01
黑龙江交通科技(2021年9期)2021-10-13
石油化工技术与经济(2020年1期)2020-12-31
石油石化绿色低碳(2020年2期)2020-12-31
