
Gitelman综合征 1 例临床特征及基因测序分析
2020-05-15 07:30:46内蒙古自治区人民医院儿科内蒙古呼和浩特010017
罕少疾病杂志 2020年3期
内蒙古自治区人民医院儿科 (内蒙古 呼和浩特 010017)
窦忠霞 马春艳 吴成君 张立泽
Gitelman综合征(Gitelman syndrome,GS),是一种罕见的常染色体隐性遗传性疾病,本文回顾分析1例GS患儿的临床资料以及患儿及其父母的基因测定结果,并通过SLCl2A3基因突变分析探讨其基因突变类型,报告如下:
1 临床资料
患儿,女,6岁,主因“发热、咽痛伴心前区疼痛3天”收住院。患儿于入院前3天无明显诱因出现发热,热峰40.0℃,无头晕头痛,无寒战及抽搐,伴咽痛,无咳嗽及喘息,伴心前区疼痛,呈钝痛,持续数分钟后自行缓解,一日出现数次,病程中无呕吐、腹泻及腹胀、腹痛,无尿频尿急及尿痛,食纳差,大小便如常。6月前因“化脓性扁桃体炎、低钾血症”在其他医院住院治疗7天。平素多饮、多尿,夜尿多,偶有肢体疲乏、无力症状。否认使用利尿剂、胰岛素及其他药物史。否认慢性腹泻、呕吐病史。否认遗传病史及家族史。查体:T38.1℃,P140次/分,R25次/分,SpO296%22kg,神清,精神可,呼吸平稳,无鼻翼扇动,咽充血明显,扁桃体II°大,可见脓性分泌物,双肺呼吸音粗,未闻及干湿啰音,心率140次/分,律齐,未闻及杂音,腹平软,无压痛、反跳痛及肌紧张,神经系统检查无异常。肌力5级,肌张力无异常。入院后血压波动在83~96/53~65mmHg之间,尿量:1990~2770ml/天,入院时心电图:交界性心动过速,部分(II、III、aVF)导联T波低平。B超:肝胆胰脾双肾、输尿管未见明显异常,双侧肾上腺未见明显异常。化验:血常规:WBC16.78×109/L,N80.9%,PLT291×109/L,RBC4.51×109/L,Hb135g/L,CRP115.99mg/L,尿常规PH7.0比重1.010,镜检(-),血生化:总蛋白77.2g/L,白蛋白53.5g/L,葡萄糖4.01mmol/L,总胆红素17.54umol/L,尿酸356 mmol/L,CK-MB5u/L,LDH271u/L,AST40u/L, ALT16 u/L,CO2CP 21.76,肌酐36.4umol/L,淀粉酶59u/L,cTnI<0.01ug/l,NT-pro-BNP132ug/l,血钾2.07mmol/L,血氯86.8mmol/L,血钠131.5mmol/L血钙2.25mmol/L,AG22.94mmol/L;2次血镁检测0.85mmol/L及0.73mmol/L(参考值0.75~1.02mmol/L),血气分析(静脉血)PH7.46,PCO236.1mmHg,PO259mmHg,HCO325.4mmol/L,SBE 1.9mmol/L。胰岛素(空腹)16.52mU/L(参考范围3.00~25.00),C肽(空腹)1.31ng/ml(参考范围0.81~3.85),皮质醇17.36ug/dl(参考范围5.27~22.457-9时)ACTH29.49pg/ml(参考范围0.00~46.00), FT33.58pg/ml(参考范围1.5~4.1), FT41.76ng/dl(参考范围0.8~1.8)TSH1.948Uiu/ml(参考范围0.4~5.0)。入院后每天补充氯化钾1.5~2.5g,血钾波动在2.07~3.66mmol/L,血氯86.8~98.7mmol/L,血钠131.5~137.3mmol/L,血钙2.25~2.54mmol/L。
入院后先后2次进行24小时尿检:第一次尿量2450ml,尿钙0.25mmol/24h,尿钾28.89mmol/24h(血钾2.79mmol/L),尿钠184.73mmol/24h,第二次24小时尿量2300ml,尿肌酐3.11mmol/24h,尿钙0.32mmol/24h,尿钾51.64mmol/24h(血钾3.13mmol/L),尿钠143.98(mmol/24h),尿氯118.45(mmol/24h)。2次检测RAAS,第一次检测:肾素活性26.82ng/ml/h(卧位0.15~2.33),醛固酮363.90pg/ml(卧位30~160),第二次检测:血管紧张素II87.25pg/ml(卧位25~60),肾素活性7.38ng/ml/h(卧位0.15~2.33),醛固酮109.5pg/ml(卧位30~160),血管紧张素II96.28pg/ml(卧位25~60)。北京海思特医学检验实验室检测患儿基因,检测内容:SLC12A3基因外显子1-26、IVS13-191、IVS21+253,用PCR和基因测序方法检测标本中SLC12A3基因编码区1-26外显子和两个剪切位点(IVS13-191、IVS21+253)变异,涵盖了该范围内的点变异、插入和缺失型变异。共进行26个PCR扩增反应和52个基因序列测定反应。该标本SLC12A3基因所检测区域共检测到7个变异位点,其中:rs1529927 Exon12错义变异c.1456G>A(p.Asp486Asn)(杂合),HGMD数据库收录的Gitelman 综合征致病性突变(CM961290)[1-2]。rs2304483,Exon16错义变异c.1964G>A(p.Arg655His)(杂合),HGMD数据库收录的Gitelman 综合征致病性突变(CM961296)[1-2]。此外还检测到Exon2同义变异c.366A>G(p.Ala122=)(纯合),Exon8同义变异c.1023C>T(p.Phe341=)(杂合)不引起氨基酸的改变,Exon6错义变异c.791C>G(p.Ala264Gly)(纯合)Exon 14内含子变异c.1670-8T>C(杂合)据目前数据库检索,该位点为SLC12A3基因的单核苷酸多态性位点,无致病性Exon23错义变异c.2738G>A(p.Arg913Gln)(杂合)意义暂不明确(rs11643718)。SLC12A3基因位于第16号染色体上,该患儿致病基因外显子测序,见图1-2。

表1 SLC12A3基因突变检测结果

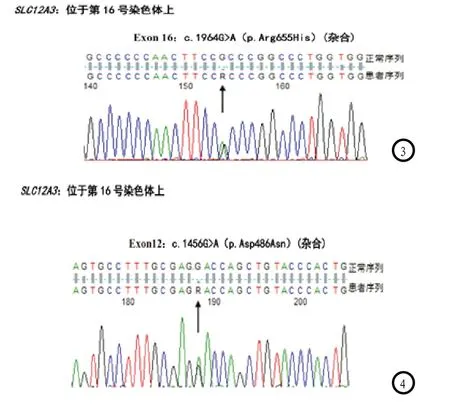
同时也检测了患儿母亲及父亲的SLC12A3基因的第12,16外显子:患儿母亲:采用PCR、测序的检测方法。检测结果:SLC12A3(NM_000339.2)Exon12无变异,Exon16错义变异c.1964G>A(p.Arg655His)(杂合),HGMD数据库收录的Gitelman 综合征致病性突变(CM961296)。患儿母亲Exon基因外显子测序图:SLC12A3 Exon16错义变异c.1964G>A(p.Arg655His)(杂合),见图3。
患儿父亲:PCR、测序的检测方法检测SLC12A3基因第12,16外显子,检测结果:(CM961290), Exon 16无变异,SLC12A3(NM_000339.2) Exon 12 c.1456G>A(p.Asp486Asn)(杂合),HGMD数据库收录的Gitelman 综合征致病性突变[1-2],见图4。患儿及其父母基因检测结果见表1。
2 讨 论
Gitelman综合征(Gitelman syndrome,GS),是一种罕见的常染色体隐性遗传性疾病,造成其编码产物噻嗪类敏感的Na+/CL一共转运蛋白(thiazide,sensitive NaCl.cotransporter,NCCT)功能失活或缺陷,最终引起疾病的发生。NCCT表达定位于肾远曲小管管腔膜上,其主要作用是促进水和Na+重吸收。典型GS患者临床表现为“五低一高”,即低血钾、低血镁、低血氯、低尿钙、偏低血压和RAAS活性增高,特别是低血镁和低尿钙对诊断GS有重要价值[3-5]。本例患儿因急性扁桃体炎就诊,因常规血液检发现严重低钾血症,进一步检查诊断本病,Fujimura[6]报道日本185例患儿通过偶然血液检测而诊断本病占54.7%。本例患儿本次就诊前半年也因呼吸道感染曾检测血钾低,当时并未重视,使患儿诊断推迟半年,文献报道GS的发病率尚不清楚,国外报道欧洲人中约为1/40000,日本人中估算的患病率在10.3/10000[3]。GS患者一般在青少年或成年时发病,血尿生化异常可早于临床症状出现。我国目前尚缺乏我国GS发病率的数据,虽然是我国首批121种罕见病之一,临床真正的发病率可能并不是很低,提高本病的临床认知度是非常必要的。
患儿存在低血钾、低血氯、低尿钙、偏低血压和代谢性碱中毒及RAAS活性增高。常规补钾治疗效果不好,先后2次血镁检测患儿血镁处于正常低限或轻度降低,低镁血症不是非常严重。2017年KDIGO[4]关于GS的争议性共识中,认为低血钾和低血镁的严重程度决定了患者临床表现的严重性,随着基因诊断金标准建立,血镁正常GS患者逐渐引起关注,文献报道[5]GS正常血镁患者比例约为8%~22%,本例患儿血镁水平仍需长期关注。
本例患儿入院时心电图提示:交界性心动过速,部分(II、III、aVF)导联T波低平。本病过去曾被认为是一种良性疾病,但严重的室性心律失常和与QT间期延长相关的心源性猝死在罕见的病例中被报道[7]。本患儿住院后经补钾治疗后心电图回复正常,心电图变化是GS疾病的表型还是低钾血症所致,还有待长期随访观察。
本例患儿SLC12A3基因所检测区域共检测到7个变异位点,其中rs1529927 Exon 12错义变异c.1456G>A(p.Asp486Asn)(杂合)及rs2304483,Exon 16错义变异c.1964G>A(p.Arg655His)(杂合)是HGMD数据库收录(CM961296)及(CM961290)的Gitelman 综合征致病性突变[6-7]。前者患儿父亲携带,后者患儿母亲携带。患儿为非单杂合突变,其父母单杂合突变携带,目前未致病。该患儿标本SLC12A3基因所检测区域还检测到其他5个变异位点:Exon2同义变异c.366A>G(p.Ala122=)(纯合),Exon8同义变异c.1023C>T(p.Phe341=)(杂合)不引起氨基酸的改变,Exon6错义变异 c.791C>G(p.Ala264Gly)(纯合)Exon14内含子变异c.1670-8T>C(杂合),Exon23错义变异c.2738G>A(p.Arg913Gln)(杂合)(rs11643718)意义暂不明确。这5个变异点或为SLC12A3基因无致病性的单核苷酸多态性位点,或为同义变异,不引起氨基酸的改变。
Balavoine等[8]研究发现大多数GS患者SLCl2A3基因检测出两个突变位点,少数患者的等位基因只检测出一个突变位点,且具有两个突变位点患者的临床症状比只发现一个突变位点的要严重。根据对该患者及其家系SLCl2A3基因突变分析得出,该患者基因变异为复合杂合突变c.1456G>A(p.Asp486Asn)和c.1964G>A(p.Arg655His),该家系共II代3例成员,其中Gs患者1例(Ⅱ1),表型正常成员2例,先证者父母为非近亲婚配,患儿父亲及母亲各发现1个突变位点,其传递方式符合孟德尔遗传法则。理论上该家系遗传特点包括:(1)先证者双亲表型正常但均为致病突变杂合子携带者,先证者的两个致病等位基因的突变性质并不相同,即分别从父方及母方传得不同致病突变,而呈突变复合杂合子。(2)同代同胞中1/4同胞发病,1/2同胞为不患病的杂合子携带者,另1/4同胞正常,男女发病机会均等。(3)患者的子女中1/2为杂合子携带者,男女相等。常染色体隐性遗传病在杂合状态时不表现相应性状,只有当一对等位基因是隐性致病基因纯合子或复合杂合子时才发病。本例患儿系复合杂合子。Gs是常染色体隐性遗传病,理论上携带者不会发病,本例家庭成员中父亲及母亲各带有一个突变基因,作为携带者目前没有发病迹象,但不能排除远期是否一定不发病,需临床关注。本例患儿父母曾有两次孕早期流产史,目前患儿母亲再次妊娠孕20周,孕期顺利。由于GS患者预后良好,妊娠期患者胎儿GS的产前诊断目前在世界范围内均未开展。临床需要密切关注,争取出生后早期诊断,早期干预,早期治疗,将病情控制在最理想范围。
Gs临床表现可不典型,在临床工作中遇到慢性低钾血症及代谢性碱中毒的患儿需考虑此病,完善基因检查,明确诊断的同时还可能会发现新的致病基因,以推进该疾病的研究。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
心电与循环(2021年4期)2021-11-29 02:41:56
种子(2021年3期)2021-04-12 01:42:22
心电与循环(2020年3期)2020-06-18 13:43:12
中国眼镜科技杂志(2016年17期)2016-10-24 08:36:30
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
浙江医学(2014年17期)2014-04-13 10:13:16
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
