
单细胞RNA测序技术在病毒研究中的应用
2020-04-15 03:16屈亮李素仇华吉
遗传 2020年3期
屈亮,李素,仇华吉
综 述
单细胞RNA测序技术在病毒研究中的应用
屈亮,李素,仇华吉
中国农业科学院哈尔滨兽医研究所,兽医生物技术国家重点实验室,哈尔滨 150069
单细胞RNA测序(single-cell RNA sequencing, scRNA-seq)技术已经成为不同领域中研究细胞异质性的有效工具。在病毒研究领域中,利用该技术分析病毒和细胞的转录组,可以在单细胞水平上检测病毒感染的动态变化,了解病毒与细胞间复杂的相互作用。本文简述了scRNA-seq技术,着重介绍病毒感染宿主细胞后scRNA-seq研究的最新进展,同时也描述了细胞周期、基因表达、细胞状态等细胞异质性对病毒感染过程的影响,以及病毒变异对其本身感染过程的影响。此外,本文还分析了scRNA-seq在研究病毒–宿主互作动态变化方面具有的独特优势,及其在病毒研究领域中广阔的应用前景,为揭示病毒的感染与致病机制、抗病毒靶标的开发等提供参考。
单细胞RNA测序技术;细胞异质性;病毒感染
单细胞测序(single-cell sequencing, SCS)技术是在单个细胞水平上测序基因组和转录组的技术,主要分为单细胞DNA测序(single cell genomic DNA sequencing)与单细胞RNA测序(single-cell RNA sequencing, scRNA-seq)。SCS技术能够区分细胞群体中单个细胞间的差异,从而反映出一些少量细胞所具有的独特表型。近年来,这些少量的具有异质性的独特细胞受到了越来越多的关注。
细胞异质性是生物组织所具有的普遍特征[1]。生物个体内的细胞最初都来自于相同的受精卵细胞。但随着细胞分裂和不同的微环境影响,细胞逐渐累积基因突变,最终形成具有不同异质性的细胞。通过SCS技术分析单个细胞的核酸数据,可以揭示是否存在稀有细胞,发现新型功能基因,探究疾病进程中的分子机制[2~4]。本文在简单介绍scRNA-seq技术的基础上,着重综述了该技术在病毒研究领域中的应用进展,旨在探索病毒多样性以及单个细胞异质性对病毒复制周期的影响,为深入揭示病毒的感染和致病机制提供科学依据。
1 单细胞RNA测序技术
细胞群体分析是研究细胞生物学和病原体感染反应的重要方法,通过分析细胞群体的转录组可以进一步发掘未知转录本。群体转录组分析技术主要包括微阵列技术以及RNA测序(RNA-seq)技术。与微阵列技术相比,RNA-seq检测范围更广,能够直接测定片段序列,是研究基因表达和鉴定新RNA种类的首选方法。然而,细胞群体含有多种细胞,这些细胞在很多方面存在差异,如细胞类型、亚群、谱系、周期、昼夜节律以及一些随机性变化,群体转录组分析结果无法评价单个细胞的转录组水平,具有特定表型的稀有细胞和亚群在群体分析中极易被忽略掉。Tang等[5]于2009年首次开发出scRNA- seq技术,该技术能够精确地检测单个细胞中的RNA分子,可用于评估细胞群体内的转录相似性及差异。如何分离单细胞是scRNA-seq技术的难点,针对单细胞的初期研究主要通过荧光激活细胞分选技术(fluorescence activated cell sorting, FACS)或延时显微镜研究携带荧光的报告病毒在单个细胞中的表达情况[6]。
1.1 单细胞RNA测序实验流程
scRNA-seq技术包括3个基本步骤:单细胞分离、RNA-seq过程和数据分析(图1)。目前单细胞分离方法可以根据两个标准划分:细胞分离能力以及细胞选择方法。细胞分离依据分离能力的不同可分为高通量和低通量分离方法,而细胞选择方法可分为随机筛选和特异性筛选,如FACS能够根据细胞大小、形状或特定的表面标志物分选细胞,微流体技术则是随机的分离单个细胞[7,8]。
单细胞分离出来后,随即开展的是RNA-seq过程。但由于一个单细胞平均仅含有约10 pg的总RNA,所以需要调整优化RNA-seq过程以适应单细胞检测。首先需要裂解分离的单细胞以获得RNA,然后通过polyA选择富集mRNA,并利用修饰过的oligo(dT)引物进行反转录,接着进行体外转录或聚合酶链式反应(polymerase chain reaction, PCR)扩增cDNA,扩增后的cDNA用于后续的测序[9]。
最后为数据分析阶段,因单细胞的RNA测序无法进行重复实验,所以对于单细胞RNA测序的数据分析必须通过质量控制保证数据的可靠性,如向细胞裂解物中添加已知序列和质量的mRNA作为质控标准[10]。
1.2 单细胞RNA测序技术的应用
目前,scRNA-seq技术已经在癌症、免疫学、细胞生物学、病毒学、胚胎学和微生物学等多个领域中得到了广泛的应用[11]。例如,在肿瘤研究方面,利用scRNA-seq可以识别肿瘤细胞之间基因突变的情况,区分瘤内细胞形成的不同类型[12],为研究肿瘤异质性及临床诊断治疗提供有效工具。在免疫学研究方面,利用scRNA-seq揭示CD4+细胞毒性T淋巴细胞在人体中的异质性和转录本,从而探究其产生机制和功能特性[13]。在细胞生物学研究方面,scRNA-seq能够更为精细地区分细胞亚群,通过分析单个细胞的转录组识别整体细胞中的各种细胞类型,可发掘已有细胞标记无法识别的新细胞类型[14]。在病毒研究方面,scRNA-seq用以区分单个细胞间病毒丰度差异,鉴定宿主细胞中的抗病毒因子,探究病毒与宿主相互作用机制[15]。在胚胎学研究中,利用scRNA-seq可分析不同分化阶段细胞之间的差异,进而解析干细胞分化过程的分子机制[16];还可以通过scRNA-seq技术可建立早期胚胎发育过程的基因表达动态图谱,探究发育过程中细胞内部的转录调控及表观遗传重编程,使得研究胚胎发育早期阶段基因表达成为可能[17]。在微生物学研究中,scRNA-seq能够显示非生长和生长中沙门氏菌()感染不同的宿主反应状态,从而揭示了沙门氏菌免疫逃逸的机制[18]。本文将重点介绍scRNA- seq技术在病毒研究中的应用。
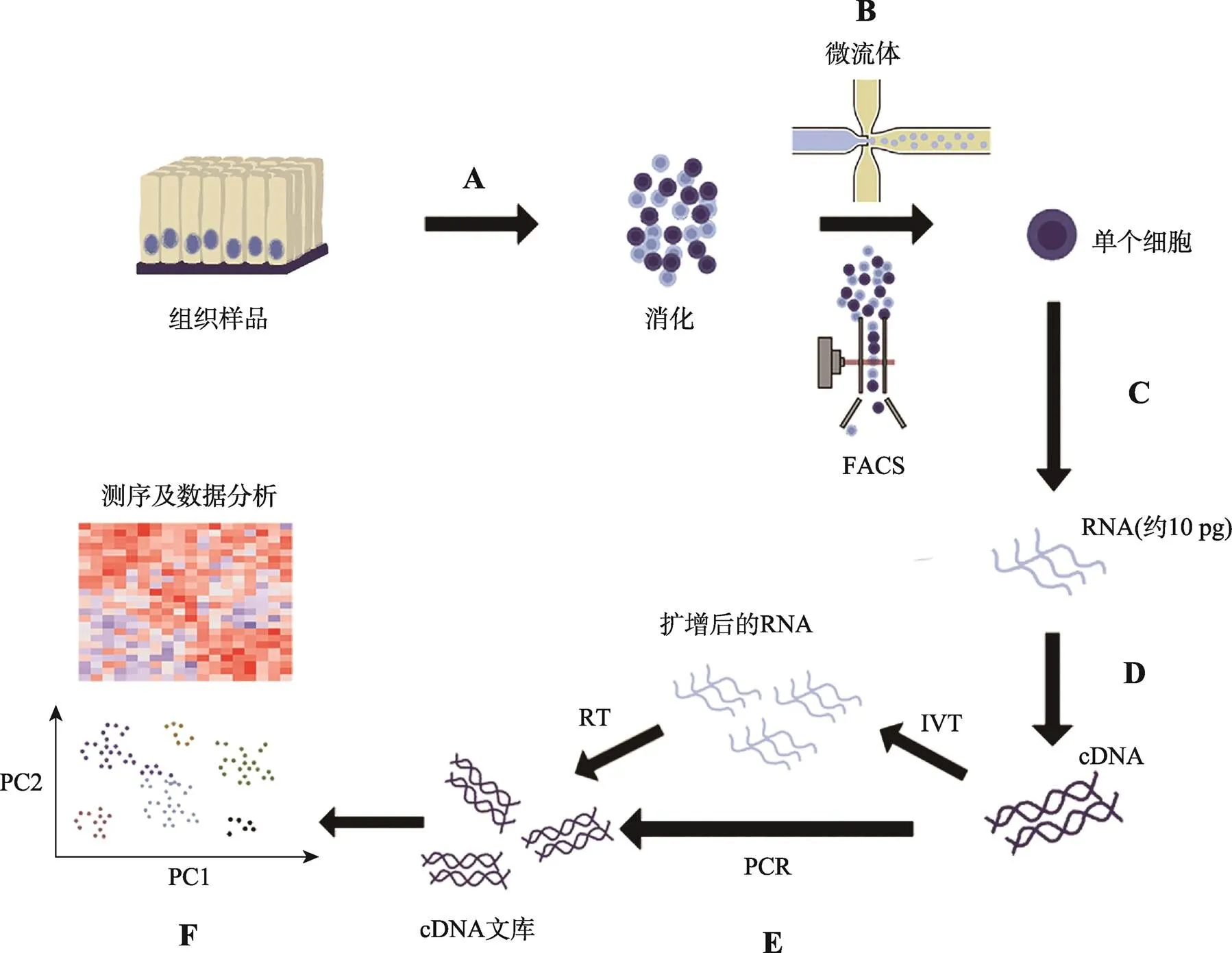
图1 单细胞RNA测序流程图
A:组织样品消化;B:微流体或FACS分离单个细胞;C:单个细胞核酸提取;D:全转录组扩增;E:体外转录及反转录或PCR扩增构建cDNA文库;F:测序数据分析。
1.3 单细胞RNA测序衍生技术
随着科学研究的不断深入,对技术手段的要求不断提高,scRNA-seq技术在许多领域中迅速优化,发展成为适合不同领域的测序技术。例如,分析冷冻固定样本的单细胞核测序技术(single nuclei RNA- sequence, sNuc-seq),突破了scRNA-seq只能分离新鲜组织样本的限制,使其在癌症研究及神经元研究中发挥着极其重要的作用[19]。sNuc-seq不需要通过蛋白酶消化和加热的方式即可快速解离样品,减少了异常转录的可能性。在sNuc- seq的基础上,有研究结合了5-乙炔基-2'-脱氧尿苷(5-ethynyl-2'-deoxyuridine, EdU)标记分裂细胞的特性,开发出了单分裂细胞核测序技术(Div-seq),能够敏感地分析中枢神经系统神经元多样性和动态过程[20]。成对单细胞RNA测序技术(paired dual scRNA-seq)能够同时分析单个宿主细胞和单个细菌之间的相互作用[21],可同时捕获和检测宿主与细菌的转录本,为微生物学研究提供重要技术支撑。
2 单细胞RNA测序技术在病毒研究中的应用
随着scRNA-seq技术的不断发展,其在病毒研究领域中的应用也得到了更进一步地提升(表1)。利用scRNA-seq技术,可以研究病毒感染后单个细胞中病毒与宿主细胞的转录组。依据病毒感染特点,分析不同状态的病毒转录组差异,探究某些病毒发生潜伏感染的基因表达情况与所需的细胞环境差异。已有研究发现,人腺病毒(human adenovirus, HAdV)潜伏感染与裂解性感染的单个细胞中病毒核酸含量有所差异,从而使得病毒E1A 13S和12S mRNA呈现出不同的表达方式[22]。另外,不同细胞对病毒的易感性不同,根据病毒感染水平的不同,分析所感染的单个细胞之间有何差异及共性。例如,单核细胞分化为巨噬细胞时其对病毒的易感性会发生变化。与单核细胞相比,巨噬细胞对于甲型流感病毒(influenza A virus, IAV)及HIV更为易感[23]。细胞对于HIV易感性的差异是由于细胞分化状态的不同引起的,也与细胞内基因表达差异有关,包括APOBEC3A与3G、CCR5、靶向HIV-1 3¢UTR的miRNA[24,25]。由于病毒与宿主之间的相互关系比较复杂,将从细胞异质性及病毒异质性对病毒感染过程的影响两方面进行介绍。

表1 scRNA-seq在病毒研究中的应用
2.1 细胞异质性影响病毒感染过程
2.1.1 细胞周期影响病毒感染
细胞周期是影响病毒感染效率的一个关键因素,许多病毒会编码靶向细胞周期调节因子的蛋白质,使宿主细胞处于有利于病毒复制的状态,如HCV和HIV感染细胞后G1期和S期的细胞数量会显著减少,但G2/M期的细胞数量会增加[33]。病毒介导的细胞周期改变虽有利于病毒生长,但对细胞本身会产生不利影响。反之,细胞大小和细胞周期的不同也会显著影响水疱性口炎病毒(vesicular stomatitis virus, VSV)子代病毒的产生数量[34]及口蹄疫病毒(foot-and-mouth disease virus, FMDV)的复制能力[35]。
为探究细胞周期对FMDV感染能力的影响,将FMDV感染后的细胞分选为大小、重量和周期相异的单个细胞,通过scRNA-seq方法对单个细胞进行分析,确定细胞间的差异以及不同细胞对FMDV感染的影响[35]。结果显示,单个FMDV感染细胞中病毒RNA数量差异达100倍,说明宿主细胞周期异质性对于FMDV复制有显著影响。在脊髓灰质炎病毒(poliovirus, PV)及IAV感染的细胞中,不同细胞中病毒RNA水平相差1~3个数量级[36,37],说明细胞周期异质性会影响多数病毒复制。对感染细胞大小及内容物进行研究,发现形态较大、含有较多细胞器的细胞中病毒蛋白及RNA含量、感染阳性率及FMDV吸附水平更高。与FMDV研究结果类似,VSV在较大的细胞中能产生更多的子代病毒[38]。多数蛋白质与mRNA含量、细胞器的大小和数量都随细胞形态变大而增加[39],所以形态较大或细胞器较多的细胞内含有更多病毒感染复制所需的物质。
部分RNA病毒会在特定的细胞周期干扰细胞从而导致细胞停滞生长。例如,HCV诱导细胞停滞于G2/M期[33],流感病毒(influenza virus, IV)使细胞停滞于G0/G1期[40],使病毒复制处于更有利的环境。FMDV感染细胞后对细胞周期没有显著影响,但不同细胞周期FMDV复制能力差异显著。G2/M期相比于其他阶段的细胞内病毒RNA含量及感染率也更高。利用药物在不同阶段阻断细胞周期,发现G2/M期停滞的细胞更有利于病毒复制以及子代病毒的产生。不仅是细胞周期,细胞的大小及细胞器含量均会对病毒感染产生显著影响,针对宿主细胞的异质性因素进行调控,找到限制病毒感染复制的优化条件。
2.1.2 病毒感染引起细胞基因表达差异
病毒感染本身会触发细胞基因表达,差异表达基因因细胞而异,并增加细胞的异质性。对HIV潜伏感染及激活后裂解性感染的细胞进行scRNA-seq分析,发现两类细胞有着显著的差异[41,42],潜伏感染的细胞大部分会表达特定的转录因子,并且在复制能力高的细胞中更易发生潜伏感染。对单纯疱疹病毒(herpes simplex virus, HSV)潜伏感染的scRNA- seq分析显示了相似的结果,HSV裂解性感染阶段的基因表达影响了宿主细胞的基因表达[43],说明裂解性感染与潜伏感染的细胞中存在细胞微环境的差异及胞内基因表达的差异。为检测病毒感染细胞差异基因表达,开发出包含病毒的单细胞RNA-seq (viscRNA-seq)[32]用于量化分析登革热患者体内单个免疫细胞的宿主mRNA以及vRNA丰度,同时还可鉴定新的抗病毒因子。该方法首先分离患者的外周血单个核细胞(peripheral blood mononuclear cell, PBMC),通过FACS将其分离为T细胞、NK细胞、B细胞、单核细胞以及树突状细胞,利用viscRNA- seq对每个细胞进行深度测序,观察每个细胞的细胞类型、免疫激活状态、vRNA水平和毒株的序列,检测到多个在登革热患者细胞中上调表达的基因,并以此作为登革热疾病严重程度的预测指标。
研究ZIKV的过程中同样利用viscRNA-seq分析其感染神经干细胞后的基因表达差异[29]。该研究在构建ZIKV-Dak-MA小鼠适应性毒株时发现,NS4B蛋白存在氨基酸突变,且突变后毒株致病性增强,通过viscRNA-seq确定这些突变如何影响ZIKV致病性。结果显示,突变毒株感染细胞内的干扰素刺激基因(interferon-stimulated gene, ISG)表达水平低于野生型毒株感染细胞,说明突变毒株有效拮抗干扰素(interferon, IFN)反应,揭示突变毒株在细胞内感染性和致病力增强的机制。此外,利用该技术研究DENV和ZIKV感染所致的差异[44],比较两者与宿主细胞相互作用有何不同。鉴定出黄病毒复制过程中所涉及的几种细胞功能,包括内质网易位、N-连接糖基化和细胞内膜运输。首先验证DENV感染后引起的差异基因表达与病毒感染的相关性,大多具有强正相关的基因均参与了非折叠蛋白反应(unfolded protein response, UPR),与前任的研究结果一致[45],而强负相关的基因很大部分是肌动蛋白和微管的组分,说明在病毒感染过程中细胞骨架遭到破坏。感染后不同时间点采样,发现在感染后4 h观察到细胞内参与翻译和抑制mRNA加工的基因上调,而在48 h,UPR上调,ERAD蛋白降解。随着感染时间的增加,有6种基因的表达从下调变为上调,部分蛋白在之前已有过报道,例如RPN1和HM13对DENV感染有很重要的作用[46],COPB1的亚基在DENV复制中发挥关键作用[47]。
2.1.3 病毒感染触发先天免疫应答
病毒入侵细胞后病原相关分子模式(pathogen- associated molecular patterns, PAMP)被胞内模式识别受体(pattern recognition receptor, PRR)所识别,产生IFN及其他细胞因子诱导先天免疫应答。但IV感染后只有部分感染细胞会激活先天免疫应答[48],利用scRNA-seq技术检测单个细胞水平IFN的诱导激活情况,发现NS1蛋白在感染细胞的过程中能够抑制IFN表达[49],缺失NS1蛋白的IV感染能检测到更高水平IFN表达,说明IV只能在部分感染细胞中诱导IFN表达。WNV感染细胞的scRNA-seq分析显示了同样的结果[50],只有少数细胞能够诱导IFN-β的有效表达,且不依赖于同一细胞内病毒RNA的丰度。此外,细胞内ISG表达与病毒RNA丰度也呈现出两种截然不同的相关性,有部分ISG的表达随病毒RNA增加而急剧下降。与前人研究一致[51],WNV能够直接或间接地拮抗IFN信号转导和JAK/STAT信号通路,以对抗细胞的抗病毒效应。然而只有少部分感染细胞触发先天免疫应答的机制仍不清楚,可能是由于随机现象或者病毒感染前细胞状态就存在一些差异。即使利用合成的先天免疫配体[52]处理细胞,也只有一部分细胞能诱导产生IFN,这种效应可能与细胞处理前的染色质状态有关[53]。
2.2 病毒异质性影响病毒感染过程
病毒具有高度的遗传变异性,该特性使其能够快速进化并适应新的环境,是病毒逃逸先天免疫,产生耐药性的关键[54]。在单细胞水平分析病毒核酸序列可直接观察其遗传多样性,并且能够量化不同时间内病毒产生的遗传变异水平。有研究结合单细胞分离技术及高通量测序分析了VSV感染细胞后的两个感染周期内其遗传多样性的变化[55],发现单个细胞中会感染一种以上的突变体病毒,且不同细胞中产生遗传变异的病毒数量变化很大,说明病毒的遗传多样性对于病毒的生存和免疫逃逸至关重要。scRNA-seq作为分析单细胞序列的有效工具,在病毒遗传多样性研究中同样发挥着巨大的作用。例如,针对HSV感染细胞的scRNA-seq分析发现单个细胞中病毒基因表达有显著差异[15]。
对于IV这种具有高突变率的病毒来说,病毒遗传多样性会影响病毒感染过程。IV已进化出抑制IFN诱导的机制,但在病毒感染细胞的过程中发生突变导致病毒粒子失去免疫逃逸机制进而诱导细胞产生先天免疫应答。Russell等[56]首次从单细胞水平研究IV的遗传缺陷对于细胞内IFN表达的影响。已有大量研究表明NS蛋白在先天免疫中发挥拮抗作 用[57],但该研究证实了缺乏NS基因或NS1蛋白发生氨基酸突变后的IV是诱导单个细胞内IFN产生的主要原因。除此之外,PB1蛋白也参与了细胞先天免疫的调控,PB1发生氨基酸突变后能够增强IFN的诱导,说明该突变能够增强先天免疫。IFN表达不是由一种因素决定的,并非所有缺乏NS基因的IV均能诱导IFN表达,说明IV感染的先天免疫由多种因素共同调控。此外,病毒遗传缺陷也不能完全解释IV感染后的细胞异质性。即使利用未突变的野生型病毒粒子感染细胞,仍有少量细胞会诱导产生IFN,并且在IFN受到抑制的细胞中也观察到了IV的免疫刺激缺陷,随机性与细胞状态也同样影响着病毒感染细胞的先天免疫应答。因此,scRNA-seq可用于研究病毒变异对于单个感染细胞先天免疫调控,利用该方法能够有效地分析病毒变异。
3 结语与展望
近10年来,scRAN-seq技术得到了迅猛的发展。许多研究人员还开创了新的scRNA-seq方法,发展出了质量控制和数据分析的新型生物信息学技术,更多的是利用该方法发现了新的生物学现象,包括新细胞类型的鉴定、预测细胞状态及基因表达以及研究随机转录的功能和意义。传统的RNA-seq技术只能分析整个细胞群体的平均值,具有异质性的少数细胞不能体现出特异性的结果,而scRNA-seq技术则可分析得到细胞异质性的结果。当然,scRNA- seq技术也存在一些挑战。相对于宿主细胞的RNA,病毒RNA的含量要少很多,并且在不同细胞类型中病毒RNA含量差异也很大,需要提高文库构建的质量以使病毒RNA检测更加可行;如何有效捕获病毒mRNA也是难点之一,尽管许多病毒会将其病毒mRNA聚腺苷酸化,但也有些病毒mRNA缺少polyA尾,需要采用新的文库构建方法来满足病毒mRNA的检测;单细胞的分离是scRNA-seq技术的关键,如何从组织中快速分离单个细胞并且完整保留其转录组也是该技术发展的一大难题;此外,该技术所需硬件的价格仍然很昂贵,且后续制备cDNA文库以及深度测序成本很高,因此,如何降低成本也是目前所面临的困难。
scRNA-seq技术拓宽了对细胞异质性的理解,认识到许多领域中稀有细胞类型的存在和重要性。宿主细胞与病毒的异质性导致病毒的感染过程有着不同的结果。宿主细胞类型的不同,决定了何种细胞易被感染,何种细胞对病毒感染产生反应;细胞状态的不同,也影响了病毒与宿主间的相互作用。例如细胞周期的差异会导致病毒复制能力及子代病毒产生能力的差异;细胞内基因表达的情况也会影响病毒感染的阶段,体现疾病的严重程度;不同状态的细胞感染病毒后的反应也不同。病毒的异质性也会影响感染宿主细胞的过程,病毒遗传多样性提供给其进化能力的同时,也可能会使其产生免疫缺陷的遗传变异,产生不同突变的病毒感染细胞后也会有不一样的结果。scRNA-seq技术为病毒感染细胞过程提供高分辨率的视角,了解细胞的保护性反应,指导新型保护措施的发展并确定病毒感染的关键分子,鉴定具有保护性免疫应答的特定细胞类型,开发更有效的疫苗。关于scRNA-seq技术的研究仍处于早期阶段,未来的scRNA-seq技术将会与更多的方法相结合,如蛋白质组学、代谢组学、表观遗传学,为全面观察单个细胞提供基础,期待未来的10年人们能够更加接近单个细胞的真实面目。
[1] Kalisky T, Blainey P, Quake SR. Genomic analysis at the single-cell level., 2011, 45: 431–445.
[2] Di Palma S, Bodenmiller B. Unraveling cell populations in tumors by single-cell mass cytometry., 2015, 31: 122–129.
[3] Yuan JD, Hegde PS, Clynes R, Foukas PG, Harari A, Kleen TO, Kvistborg P, Maccalli C, Maecker HT, Page DB, Robins H, Song WR, Stack EC, Wang EN, Whiteside TL, Zhao YD, Zwierzina H, Butterfield LH, Fox BA. Novel technologies and emerging biomarkers for personalized cancer immunotherapy., 2016, 4(1): 3.
[4] Winter DR, Ledergor G, Amit I. From mass cytometry to cancer prognosis. Nat Biotechnol, 2015, 33(9): 931–932.
[5] Tang FC, Barbacioru C, Wang YZ, Nordman E, Lee C, Xu NL, Wang XH, Bodeau J, Tuch BB, Siddiqui A, Lao KQ, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell., 2009, 6(5): 377–382.
[6] Razooky BS, Gutierrez E, Terry VH, Spina CA, Groisman A, Weinberger LS. Microwell devices with finger-like channels for long-term imaging of HIV-1 expression kinetics in primary human lymphocytes., 2012, 12(21): 4305–4312.
[7] Hu P, Zhang WH, Xin HB, Deng G. Single cell isolation and analysis., 2016, 4: 116.
[8] Prakadan SM, Shalek AK, Weitz DA. Scaling by shrinking: empowering single-cell 'omics' with microfluidic devices., 2017, 18(6): 345–361.
[9] Yao YX, La YF, Di R, Liu QY, Hu WP, Wang XY, Chu MX. Comparison of different single cell whole genome amplification methods and MALBAC applications in assisted reproduction., 2018, 40(8): 620–631.姚雅馨, 喇永富, 狄冉, 刘秋月, 胡文萍, 王翔宇, 储明星. 不同单细胞全基因组扩增方法的比较及MALBAC在辅助生殖中的应用. 遗传, 2018, 40(8): 620–631.
[10] Stoeger T, Battich N, Pelkmans L. Passive noise filtering by cellular compartmentalization., 2016, 164(6): 1151–1161.
[11] Wang Y, Navin NE. Advances and applications of single- cell sequencing technologies., 2015, 58(4): 598– 609.
[12] Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, Rodman C, Luo CL, Mroz EA, Emerick KS, Deschler DG, Varvares MA, Mylvaganam R, Rozenblatt- Rosen O, Rocco JW, Faquin WC, Lin DT, Regev A, Bernstein BE. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer., 2017, 171(7): 1611–1624.
[13] Patil VS, Madrigal A, Schmiedel BJ, Clarke J, O'rourke P, De Silva AD, Harris E, Peters B, Seumois G, Weiskopf D, Sette A, Vijayanand P. Precursors of human CD4+cytotoxic T lymphocytes identified by single-cell transcriptome analysis., 2018, 3(19): eaan8664.
[14] Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single-cell rna-seq for marker-free decomposition of tissues into cell types., 2014, 343(6172): 776–779.
[15] Drayman N, Patel P, Vistain L, Tay S. HSV-1 single-cell analysis reveals the activation of anti-viral and developmental programs in distinct sub-populations., 2019, 8: e46339.
[16] Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells., 2014, 32(4): 381–386.
[17] Yan LY, Yang MY, Guo HS, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng XY, Yan J, Huang J, Li M, Wu XL, Wen L, Lao KQ, Li RQ, Qiao J, Tang FC. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells., 2013, 20(9): 1131–1139.
[18] Saliba AE, Li L, Westermann AJ, Appenzeller S, Stapels DA, Schulte LN, Helaine S, Vogel J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella., 2016, 2: 1–8.
[19] Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, Vijayaraghavan R, Wong J, Chen A, Sheng X, Kaper F, Shen R, Ronaghi M, Fan JB, Wang W, Chun J, Zhang K. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain., 2016, 352(6293): 1586–1590.
[20] Habib N, Li YQ, Heidenreich M, Swiech L, Avraham- Davidi I, Trombetta JJ, Hession C, Zhang F, Regev A. Div-Seq: single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons., 2016, 353(6302): 925–928.
[21] Wesolowska-Andersen A, Everman JL, Davidson R, Rios C, Herrin R, Eng C, Janssen WJ, Liu AH, Oh SS, Kumar R, Fingerlin TE, Rodriguez-Santana J, Burchard EG, Seibold MA. Dual RNA-seq reveals viral infections in asthmatic children without respiratory illness which are associated with changes in the airway transcriptome., 2017, 18(1): 12.
[22] Krzywkowski T, Ciftci S, Assadian F, Nilsson M, Punga T. Simultaneous single-cell in situ analysis of human adenovirus type 5 dna and mrna expression patterns in lytic and persistent infection., 2017, 91(11): e00166-17.
[23] Hoeve MA, Nash AA, Jackson D, Randall RE, Dransfield I. Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation., 2012, 7(1): e29443.
[24] Mussil B, Suspène R, Caval V, Durandy A, Wain-Hobson S, Vartanian JP. Genotoxic stress increases cytoplasmic mitochondrial DNA editing by human APOBEC3 mutator enzymes at a single cell level., 2019, 9(1): 3109.
[25] Lodge R, Gilmore JC, Ferreira Barbosa JA, Lombard- Vadnais F, Cohen ÉA. Regulation of CD4 receptor and HIV-1 entry by microRNAs-221 and -222 during differentiation of THP-1 cells., 2017, 10(1): 13.
[26] Afik S, Yates KB, Bi K, Darko S, Godec J, Gerdemann U, Swadling L, Douek DC, Klenerman P, Barnes EJ, Sharpe AH, Haining WN, Yosef N. Targeted reconstruction of T cell receptor sequence from single cell RNA-seq links CDR3 length to T cell differentiation state., 2017, 45(16): e148.
[27] Wu L, Zhang XL, Zhao ZK, Wang L, Li B, Li GB, Dean M, Yu QC, Wang YH, Lin XX, Rao WJ, Mei ZL, Li Y, Jiang RZ, Yang H, Li FQ, Xie GY, Xu LQ, Wu K, Zhang J, Chen JH, Wang T, Kristiansen K, Zhang XQ, Li YR, Yang HM, Wang J, Hou Y, Xu X. Full-length single-cell RNA-seq applied to a viral human cancer: applications to HPV expression and splicing analysis in HeLa S3 cells., 2015, 4(1): 51.
[28] Tsioris K, Gupta NT, Ogunniyi AO, Zimnisky RM, Qian F, Yao Y, Wang X, Stern JN, Chari R, Briggs AW, Clouser CR, Vigneault F, Church GM, Garcia MN, Murray KO, Montgomery RR, Kleinstein SH, Love JC. Neutralizing antibodies against West Nile virus identified directly from human B cells by single-cell analysis and next generation sequencing., 2015, 7(12): 1587–1597.
[29] Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger- Lucarelli J, Uccellini MB, Tripathi S, Morrison J, Yount BL, Dinnon KH 3rd, Rückert C, Young MC, Zhu Z, Robertson SJ, Mcnally KL, Ye J, Cao B, Mysorekar IU, Ebel GD, Baric RS, Best SM, Artyomov MN, Garcia- Sastre A, Diamond MS. An immunocompetent mouse model of zika virus infection., 2018, 23(5): 672–685.
[30] Johnson TS, Abrams ZB, Mo XK, Zhang Y, Huang K. Lack of human cytomegalovirus expression in single cells from glioblastoma tumors and cell lines., 2017, 23(5): 671–678.
[31] Golumbeanu M, Cristinelli S, Rato S, Munoz M, Cavassini M, Beerenwinkel N, Ciuffi A. Single-Cell RNA-Seq Reveals transcriptional heterogeneity in latent and reactivated hiv-infected cells., 2018, 23(4): 942–950.
[32] Zanini F, Robinson ML, Croote D, Sahoo MK, Sanz AM, Ortiz-Lasso E, Albornoz LL, Rosso F, Montoya JG, Goo L, Pinsky BA, Quake SR, Einav S. Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue., 2018, 115(52): E12363–E21369.
[33] Kannan RP, Hensley LL, Evers LE, Lemon SM, Mcgivern DR. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis., 2011, 85(16): 7989–8001.
[34] Timm A, Yin J. Kinetics of virus production from single cells., 2012, 424(1): 11–17.
[35] Xin X, Wang HL, Han LL, Wang MZ, Fang H, Hao Y, Li JD, Zhang H, Zheng CY, Shen C. Single-cell analysis of the impact of host cell heterogeneity on infection with foot-and-mouth disease virus., 2018, 92(9): e00179–18.
[36] Schulte MB, Andino R. Single-cell analysis uncovers extensive biological noise in poliovirus replication., 2014, 88(11): 6205–6212.
[37] Heldt FS, Kupke SY, Dorl S, Reichl U, Frensing T. Single-cell analysis and stochastic modelling unveil large cell-to-cell variability in influenza A virus infection., 2015, 6: 8938.
[38] Zhu Y, Yongky A, Yin J. Growth of an RNA virus in single cells reveals a broad fitness distribution., 2009, 385(1): 39–46.
[39] Kempe H, Schwabe A, Crémazy F, Verschure PJ, Bruggeman FJ. The volumes and transcript counts of single cells reveal concentration homeostasis and capture biological noise., 2015, 26(4): 797–804.
[40] He Y, Xu K, Keiner B, Zhou JF, Czudai V, Li TX, Chen Z, Liu JH, Klenk HD, Shu YL, Sun B. Influenza A virus replication induces cell cycle arrest in G0/G1phase., 2010, 84(24): 12832–12840.
[41] Bradley T, Ferrari G, Haynes BF, Margolis DM, Browne EP. Single-cell analysis of quiescent HIV infection reveals host transcriptional profiles that regulate proviral latency., 2018, 25(1): 107–117..
[42] Golumbeanu M, Cristinelli S, Rato S, Munoz M, Cavassini M, Beerenwinkel N, Ciuffi A. Single-cell RNA-seq reveals transcriptional heterogeneity in latent and reactivated HIV-infected cells., 2018, 23(4): 942–950.
[43] Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC. Lytic gene expression is frequent in HSV-1 latent infection and correlates with the engagement of a cell-intrinsic transcriptional response., 2014, 10(7): e1004237.
[44] Zanini F, Pu SY, Bekerman E, Einav S, Quake SR. Single-cell transcriptional dynamics of flavivirus infection., 2018, 7: e32942.
[45] Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, Defilippis V, Früh K, Mason PW, Nikolich-Zugich J, Nelson JA. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis., 2007, 81(20): 10849–10860.
[46] Marceau CD, Puschnik AS, Majzoub K, Ooi YS, Brewer SM, Fuchs G, Swaminathan K, Mata MA, Elias JE, Sarnow P, Carette JE. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens., 2016, 535(7610): 159–163.
[47] Metz P, Chiramel A, Chatel-Chaix L, Alvisi G, Bankhead P, Mora-Rodriguez R, Long G, Hamacher-Brady A, Brady NR, Bartenschlager R. Dengue virus inhibition of autophagic flux and dependency of viral replication on proteasomal degradation of the autophagy receptor p62., 2015, 89(15): 8026–8041.
[48] Killip MJ, Jackson D, Pérez-Cidoncha M, Fodor E, Randall RE. Single-cell studies of IFN-β promoter activation by wild-type and NS1-defective influenza A viruses., 2017, 98(3): 357–363.
[49] Jackson D, Killip MJ, Galloway CS, Russell RJ, Randall RE. Loss of function of the influenza A virus NS1 protein promotes apoptosis but this is not due to a failure to activate phosphatidylinositol 3-kinase (PI3K)., 2010, 396(1): 94–105.
[50] O'neal JT, Upadhyay AA, Wolabaugh A, Patel NB, Bosinger SE, Suthar MS. West Nile virus-inclusive single-cell RNA sequencing reveals heterogeneity in the type i interferon response within single cells., 2019, 93(6): e01778–18.
[51] Quicke KM, Suthar MS. The innate immune playbook for restricting West Nile virus infection., 2013, 5(11): 2643–2658.
[52] Wimmers F, Subedi N, Van Buuringen N, Heister D, Vivie J, Beeren-Reinieren I, Woestenenk R, Dolstra H, Piruska A, Jacobs JFM, Van Oudenaarden A, Figdor CG, Huck WTS, De Vries IJM, Tel J. Single-cell analysis reveals that stochasticity and paracrine signaling control interferon- alpha production by plasmacytoid dendritic cells., 2018, 9(1): 3317.
[53] Bhushal S, Wolfsmüller M, Selvakumar TA, Kemper L, Wirth D, Hornef MW, Hauser H, Köster M. Cell polarization and epigenetic status shape the heterogeneous response to type III interferons in intestinal epithelial cells., 2017, 8: 671.
[54] Lauring AS, Frydman J, Andino R. The role of mutational robustness in RNA virus evolution., 2013, 11(5): 327–336.
[55] Combe M, Garijo R, Geller R, Cuevas JM, Sanjuán R. Single-cell analysis of RNA virus infection identifies multiple genetically diverse viral genomes within single infectious units., 2015, 18(4): 424–432.
[56] Russell AB, Elshina E, Kowalsky JR, Te Velthuis AJW, Bloom JD. Single-cell virus sequencing of influenza infections that trigger innate immunity., 2019, 93(14): e00500–19.
[57] Hale BG, Randall RE, Ortín J, Jackson D. The multifunctional NS1 protein of influenza A viruses., 2008, 89: 2359–2376.
Applications of single-cell RNA sequencing in virology
Liang Qu, Su Li, Huaji Qiu
Single-cell RNA sequencing (scRNA-seq) is now emerging as a powerful tool to characterize the roles of cell heterogeneity in many fields, including virus infection. Transcriptional profiling at the single-cell level has enabled a greater appreciation of the dynamic changes of virus infection and the complex interactions between viruses and host cells. In this review, we briefly introduce the scRNA-seq technology, and the researches progress of scRNA-seq applied to virus infection. Moreover, we summarize the effects of cell heterogeneity, such as cell cycle, gene expression, and cell state, and virus mutations on the virus infection. We also analyze the unique advantages of scRNA-seq in researches of the dynamic changes of virus-host interaction, and the profound prospects of this technology used in virology for future studies. This review aims to provide a useful reference for the application of scRNA-seq in the understanding of the viral infection and pathogenicity mechanisms which may lead to the development of potential antiviral targets.
single cell RNA sequencing; heterogeneity; virus infection
2019-10-31;
2020-01-10
国家自然科学基金项目(编号:31630080,31672537)资助[Supported by the National Natural Science Foundation of China (Nos.31630080, 31672537)]
屈亮,硕士研究生,专业方向:兽医学。E-mail: tierno831143@outlook.com
仇华吉,博士,研究员,研究方向:动物疫苗与分子免疫学。E-mail: qiuhuaji@caas.cn
10.16288/j.yczz.19-223
2020/1/17 17:18:00
URI: http://kns.cnki.net/kcms/detail/11.1913.r.20200117.0949.002.html
(责任编委: 岑山)
猜你喜欢
中华骨与关节外科杂志(2022年1期)2022-08-31
中老年保健(2022年1期)2022-08-17
昆明医科大学学报(2021年8期)2021-08-13
科学(2020年4期)2020-11-26
科学(2020年3期)2020-11-26
教育界·上旬(2020年8期)2020-06-27
当代水产(2020年3期)2020-06-15
兽医导刊(2019年1期)2019-02-21
猪业科学(2018年8期)2018-09-28
江苏农业科学(2017年16期)2017-10-27
