
多组学关联分析揭示表观等位基因在拟南芥环境适应性进化中的作用及机制
2020-04-15 03:19梅志超位竹君于佳慧冀凤丹解莉楠
遗传 2020年3期
梅志超,位竹君,于佳慧,冀凤丹,解莉楠
研究报告
多组学关联分析揭示表观等位基因在拟南芥环境适应性进化中的作用及机制
梅志超,位竹君,于佳慧,冀凤丹,解莉楠
东北林业大学生命科学学院,哈尔滨 150040
表观等位基因一般是指仅由DNA甲基化差异引起的表达量不同的等位基因,对植物形态结构和各种生理过程具有重要影响。但自然条件下环境因素对植物表观等位基因的影响还不清楚,同时表观等位基因在植物环境适应性进化中的作用和机制还亟待探究。为了在全基组水平鉴定拟南芥()中与特定环境因素相关的表观等位基因,并分析它们参与拟南芥环境适应性进化的可能机制,本研究利用623株拟南芥生态型的转录组、甲基化组和种源地气候数据进行多组学关联分析,并同时进行了蛋白互作网络和基因富集分析。以春季和夏季降水量为例,本研究最终鉴定到5个基因(、、、和)可能具有相应的表观等位基因,基因内部或附近特定区域不同甲基化水平对它们的表达可能具有调控作用。其中与种子发育有关的印记基因首次被发现可能作为表观等位基因参与拟南芥环境适应性进化,其他4个基因均与生物胁迫响应有关。自然条件下降水量能影响当地病虫害的严重程度,而DNA甲基化能通过影响这4个免疫基因的表达来影响拟南芥免疫能力。在长期演化过程中有利于个体适应当地降水模式的表观等位基因受到正向选择,这可能是这些表观等位基因参与拟南芥降水适应性进化的潜在机制。通过蛋白互作网络、GO功能分析和KEGG通路分析,本研究还首次发现可能与LSU基因家族其他成员一样参与硫代谢网络,并通过影响硫代葡萄糖苷代谢参与拟南芥生物胁迫响应。
植物生态表观遗传学;多组学关联分析;表观等位基因;环境适应性进化;拟南芥
生态表观遗传学(ecological epigenetics)是由生态学和表观遗传学结合而产生的一门新兴交叉学科,主要利用表观遗传学的理论和手段分析物种演化过程和生态学研究中的一些重要现象和问题(如表型可塑性、生境分化、物种形成等),从而最终解析表观遗传学是否参与及通过何种方式参与物种(尤其植物)的长期进化与环境适应等问题[1]。DNA胞嘧啶甲基化是最常见的表观遗传标记,也是目前所有表观遗传标记中研究最透彻的。与哺乳动物胚胎期DNA甲基化重编程不同,植物细胞中有多种机制保证DNA甲基化在很大程度上稳定遗传[2]。顺式作用元件内部或附近的DNA甲基化能通过影响染色体结构、反式作用因子亲和性等调控基因转录,处于基因内部的DNA甲基化也可能通过影响转录延伸、前体RNA加工等影响基因表达水平[3]。一般将仅由于基因组特定区域DNA甲基化差异引起表达水平改变的等位基因称为表观等位基因(epiallele)[4],近年来大量表观等位基因相继被发现,揭示了表观等位基因在植物发育和逆境响应中的重要作用[5~8]。何力等[9]鉴定到一个与拟南芥()温度适应相关的表观等位基因,首次发现表观等位基因可能参与植物环境适应性进化。目前表观等位基因的鉴定手段主要是经典遗传学方法(如图位克隆),巨大的筛选工作量限制了表观等位基因的鉴定。
本研究利用“拟南芥1001项目”[10]测序的623个拟南芥生态型甲基化组、转录组和种源地气候数据进行多组学关联分析,结合蛋白互作网络分析、GO功能分析和KEGG通路分析,尝试在全基因组范围内挖掘与特定气候因素相关的表观等位基因,并探究这些表观等位基因可能的作用机制。以春季和夏季降水量为例,本研究共挖掘到5个可能参与拟南芥对当地降水量适应的表观等位基因。这些表观等位基因除之外都与拟南芥生物胁迫响应有关,因此推测当地不同降水量引起的病虫害水平差异可能是对拟南芥产生选择压力的直接因素,而这些表观等位基因通过影响拟南芥对病虫害的抵御能力参与拟南芥对当地不同降水量的适应。是受表观遗传因素调控的印记基因[11],与种子发育有关[12],本研究首次发现该基因可能作为表观等位基因参与拟南芥水分适应性进化。本研究还发现LSU (response to low sulfur)基因家族成员参与了拟南芥硫代谢网络,并可能通过调控硫代葡萄糖苷的生物合成影响拟南芥对生物胁迫的抵御能力,进而参与拟南芥水分适应性进化。
1 材料与方法
1.1 数据采集与关联分析
从“拟南芥1001”项目数据库[10]下载所有基于Illumina平台测序的拟南芥生态型甲基化组和转录组数据,GEO号分别为GSE43857和GSE80744。从AraCLIM (https://github.com/CLIMtools/AraCLIM)[13]下载所有生态型种源地春季和夏季降水量数据。选取同时具有甲基化组、转录组和降水量数据的623个生态型作为本研究的数据来源。由于甲基化对基因表达的调控往往不是通过单个胞嘧啶位点甲基化的形式,而是以一个区域内多个胞嘧啶甲基化的区域甲基化形式发挥作用,同时考虑到单碱基突变对甲基化水平的影响,本研究以Col-0基因组为参考将拟南芥全基因组划分为每200 bp为一个单位(bin),以一个bin中重亚硫酸氢盐测序所得的甲基化read数除以总read数衡量此bin甲基化水平。植物的DNA甲基化有3种序列形式,CG、CHG和CHH,但在本研究中并没有对此做出区分,因为在考察现已鉴定到的植物表观等位基因后,本研究发现3种甲基化的存在和对基因表达的调控作用没有显著差别[5~8]。使用R语言软件包Matrix eQTL[14]基于一般线性回归模型分别进行bin与基因表达关联分析(eQTLepi)、基因表达与春季/夏季降水量关联分析、bin与春季/夏季降水量关联分析(EWAS) (图1)。采用检验(student'stest)评估关联的显著性,使用FDR (false discovery rate)进行多重检验校正降低假阳性,去除FDR>0.01的结果。
1.2 表观等位基因挖掘
利用以上关联分析结果,分别选取与春季或夏季降水量相关分析值最小的50个bin用于表观等位基因挖掘的后续分析,具体分析流程见图1。提取这100个bin中至少有1个位于内部或附近(上下游2 kb以内)的基因,且要求对应bin的甲基化与该基因表达量相关。再从这些基因中筛选与春季或夏季降水量也相关的基因,即为潜在的与拟南芥降水适应相关的表观等位基因。值得注意的是,甲基化可能通过增强子或改变染色体结构远程调控基因表达,但依赖现有研究成果和数据还难以准确预测。因此,本研究中只关注基因内部或附近甲基化对基因表达的调控,而不考虑甲基化远程调控基因表达的情况。
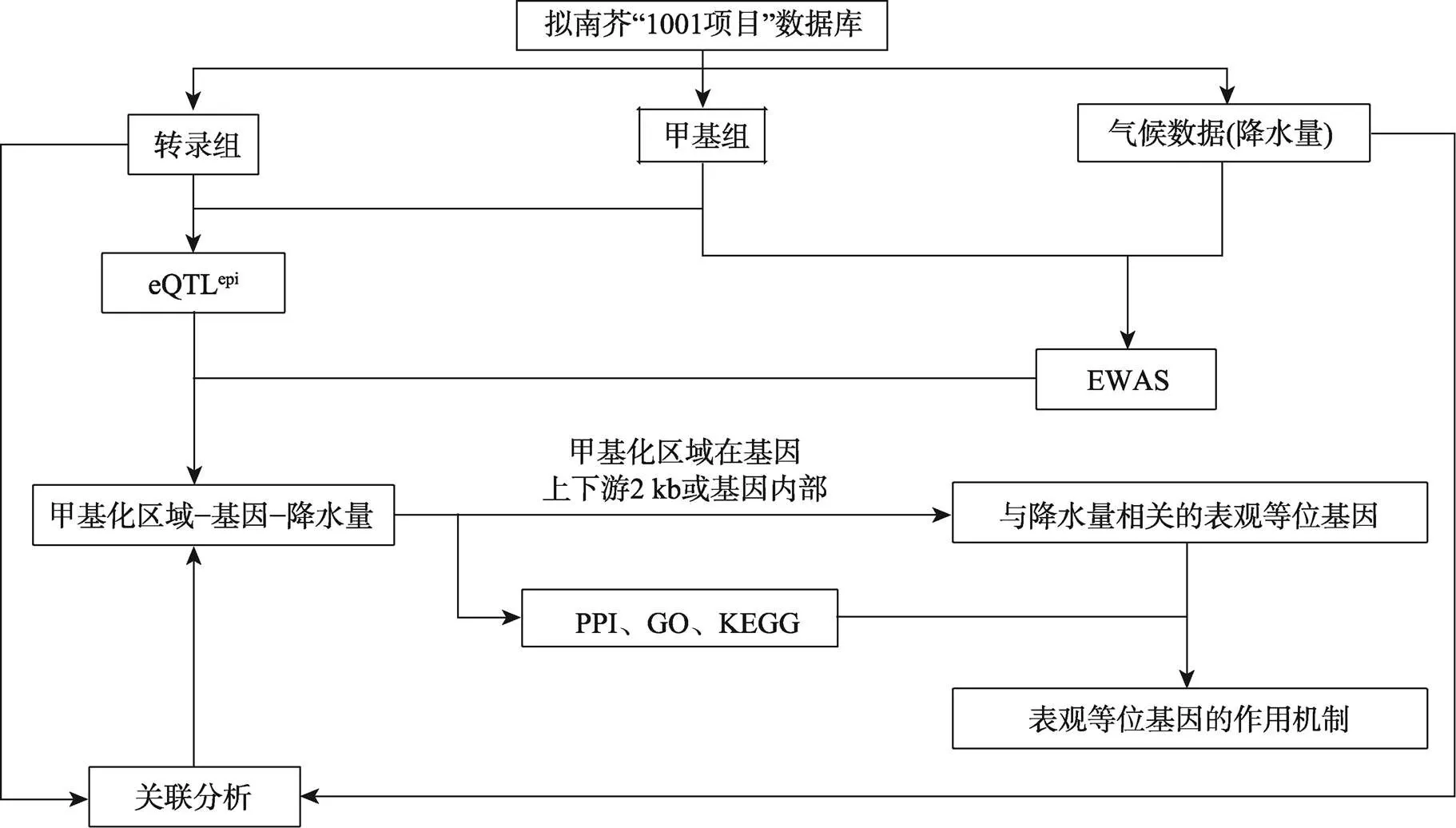
图1 本研究分析流程
通过eQTLepi(methylation-dependent eQTL)、EWAS (epigenome-wide association study)和转录组–降水量关联分析获得甲基化位点、基因、降水量两两关联的结果,再筛选甲基化位点在基因附近(上游2 kb、内部和下游2 kb)的结果,即获得潜在的表观等位基因,对所有与此甲基化位点关联的基因进行蛋白互作网络(protein-protein interaction, PPI)分析、GO注释富集分析和KEGG通路富集分析,最终获得表观等位基因潜在的作用机制。
1.3 表观等位基因作用机制分析
采用蛋白互作网络、GO和KEGG富集分析进一步挖掘甲基化通过调控基因表达影响拟南芥表型,最终参与拟南芥环境适应的具体机制。对每一甲基化区域取表达量与其甲基化水平相关的所有基因,再从这些基因中筛选与降水量也相关的基因,最终获得每一个表观等位基因对应的“基因集”。利用STRING (https://string-db.org/)对每一基因集进行蛋白互作网络分析,并使用Cytoscape[15]进行互作网络可视化。采用omicshare (https://www.omicshare.com/ tools/)在线分析工具分别对每一基因集进行GO功能分析和KEGG通路分析。根据分析结果推断这些表观等位基因是否及如何通过蛋白互作网络影响了某些代谢通路,进而影响拟南芥相关生理功能,最终影响拟南芥对春季或夏季不同降水模式的适应。
2 结果与分析
2.1 与春季降水量相关的表观等位基因
经甲基化组、转录组和春季降水数据两两关联分析和结果筛选,本研究共鉴定到两个(和)与春季降水量相关的表观等位基因(表1,图2A)。
bin01位于内部(图3A),甲基化水平与基因表达量、基因表达量与春季降水量、春季降水量与甲基化水平均呈显著正相关(图3F)。说明春季低降水量对bin01的低甲基化水平可能具有正向选择,bin01的低甲基化可能抑制表达,而该基因的高表达可能不利于拟南芥对春季低降水量的适应。参与拟南芥无义介导的mRNA降解途径(nonsense-mediated mRNA decay, NMD)。NMD是真核细胞中一种重要的RNA监控系统,能识别并降解含有提前终止密码子的mRNA,避免因截短的蛋白产物积累而对细胞造成毒害[16]。同时NMD能特异性降解植物免疫受体的转录产物,当植物受到病虫害侵袭时NMD的活动受到抑制,胞内转录产物得到积累,启动植物免疫反应[17]。降水量是影响很多病原菌侵染、繁殖、扩散和害虫种群数量、迁入迁出率的主要原因之一[18,19]。在春季低降水量的气候条件下,病虫害发生水平较高,具有bin01高甲基化的植株表达量较高,NMD对的降解作用较强,不利于植物及时启动免疫反应,最终造成这些植株逐渐被自然选择淘汰(图4)。
bin02–08位于基因内部(图3B),甲基化水平与基因表达量呈显著负相关,与春季降水量呈显著正相关,而该基因表达量与春季降水量呈负相关(图3G)。属于拟南芥免疫受体家族,参与拟南芥对病虫害的免疫反应[20]。因此本研究推测,该表观等位基因可能与的表观等位基因一样直接受当地病虫害发生水平的选择压力。bin02–08低甲基化的植株中具有较高的表达量,因此免疫能力相对较强。在春季低降水量引起的较高病虫害气候条件下,这样的植株具有更强的适应能力,在自然选择中更易被保留(图4)。
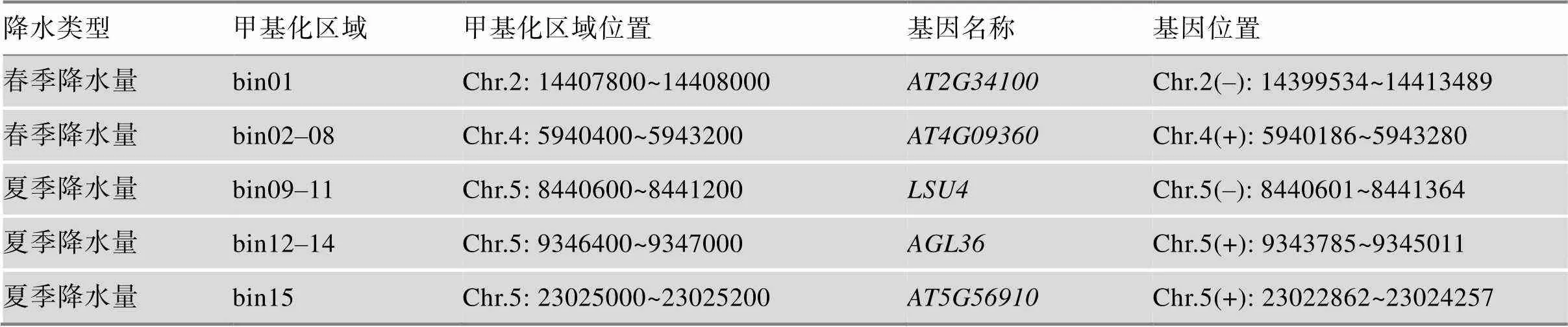
表1 与降水量相关的表观等位基因
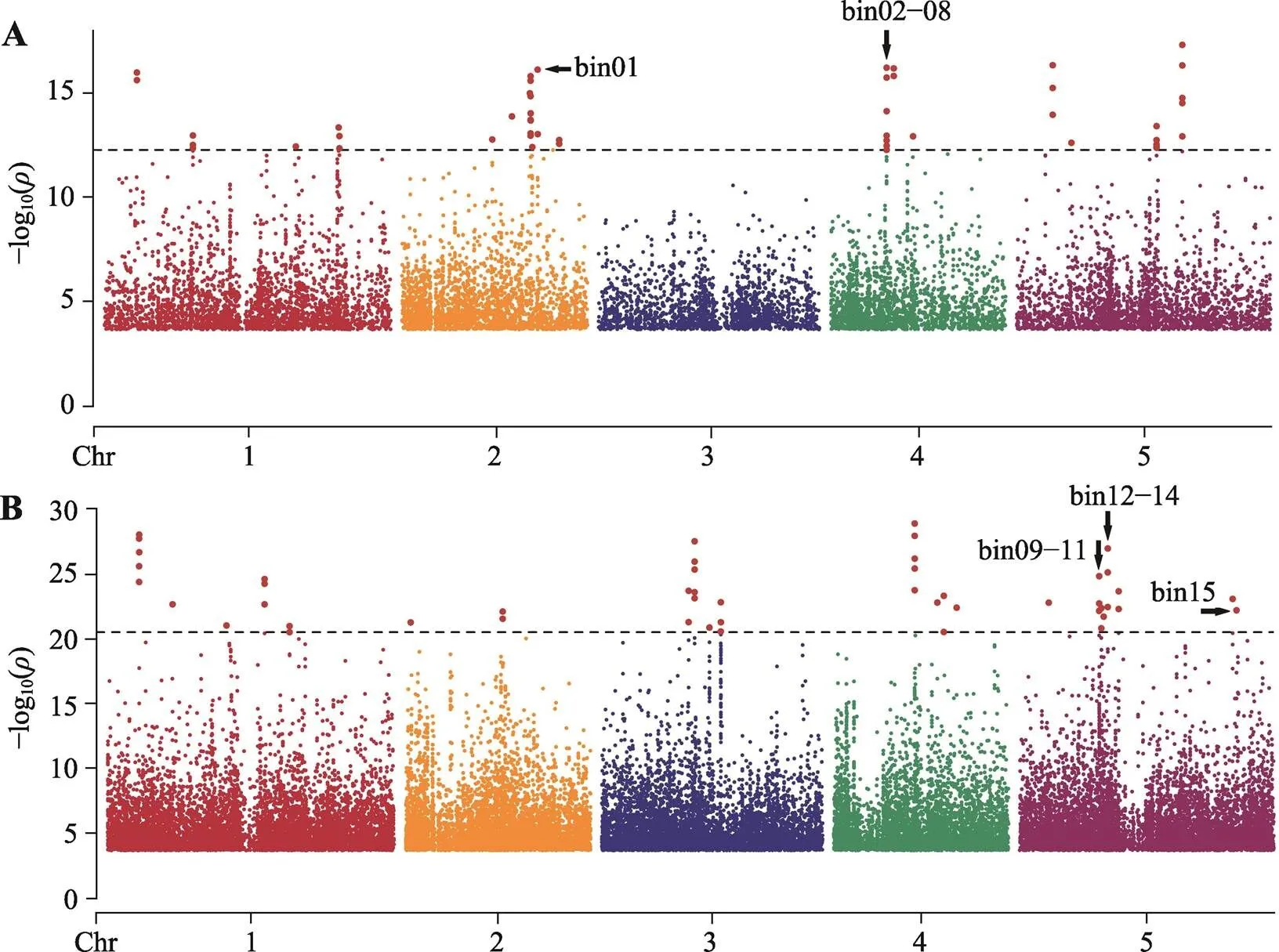
图2 曼哈顿图展示EWAS分析结果
A:春季降水量全甲基化组关联分析。虚线以上红点表示值最小的50个bin,黑色箭头标注本研究鉴定到的2个与春季降水量相关的表观等位基因对应的甲基化区域。B:夏季降水量全甲基化组关联分析。黑色箭头标注本研究鉴定到的3个与夏季降水量相关的表观等位基因对应的甲基化区域。
2.2 与夏季降水量相关的表观等位基因
经甲基化组、转录组和夏季降水量数据两两关联分析和结果筛选,获得3个(、和)与夏季降水量相关的表观等位基因(表1,图2B)。
bin09–11位于基因内部(图3C),bin12–14位于(AGAMOUS-like 36)基因下游1400 bp左右(图3D)。这两个甲基化区域的甲基化水平分别与对应基因的表达量负相关,与夏季降水量正相关,而两个基因的表达量均与夏季降水量负相关(图3,H和I)。拟南芥中LSU基因家族共有4个成员,其他3个成员均参与拟南芥低硫胁迫响应,是拟南芥硫代谢途径的关键基因[21],但目前还没有研究阐明是否参与拟南芥硫代谢途径。编码蛋白为I型MADS-box转录因子,有研究表明是与种子发育相关的印记基因,可能参与胚乳从合胞体向细胞化的发育转化,但其在种子发育中的具体作用和机制尚不清楚[11,12]。种子发育的不同模式对拟南芥适应不同夏季降水条件具有重要影响,本研究推测bin12–14的甲基能抑制的表达,不利于拟南芥对夏季低降水量的适应,在夏季低降水量的气候条件下受到净化选择(图4)。
bin15位于下游740 bp左右(图3E),甲基化水平与该基因表达量、该基因表达量与夏季降水量、夏季降水量与甲基化水平均呈显著正相关(图3J)。说明夏季低降水量可能对bin15的低甲基化有正向选择,其原因可能是bin15的低甲基化抑制表达,而该基因的高表达不利于拟南芥对夏季低降水量的适应。编码的蛋白具有半胱氨酸蛋白酶抑制剂(cysteine proteinase inhibitor, CPI)保守结构域,可能对半胱氨酸蛋白酶(cysteine proteinase, CP)具有抑制作用。CP通过促进蛋白质的降解参与植物组织衰老和细胞编程性死亡,在植物受病虫害侵袭时启动的局部超敏反应中起重要作用[22]。降低CPI的积累水平能提高CP的活性,促进受损组织的衰老和程序性死亡,限制病原体进一步扩散,提高植物对病虫害的抵御能力。因此,本研究推测受到的直接选择压力可能也是当地病虫害的发生水平,具有bin15低甲基化的植株表达水平较低,胞内CP活性较高,受病虫侵袭时局部超敏反应较强,在夏季低降水条件下受到正向选择(图4)。
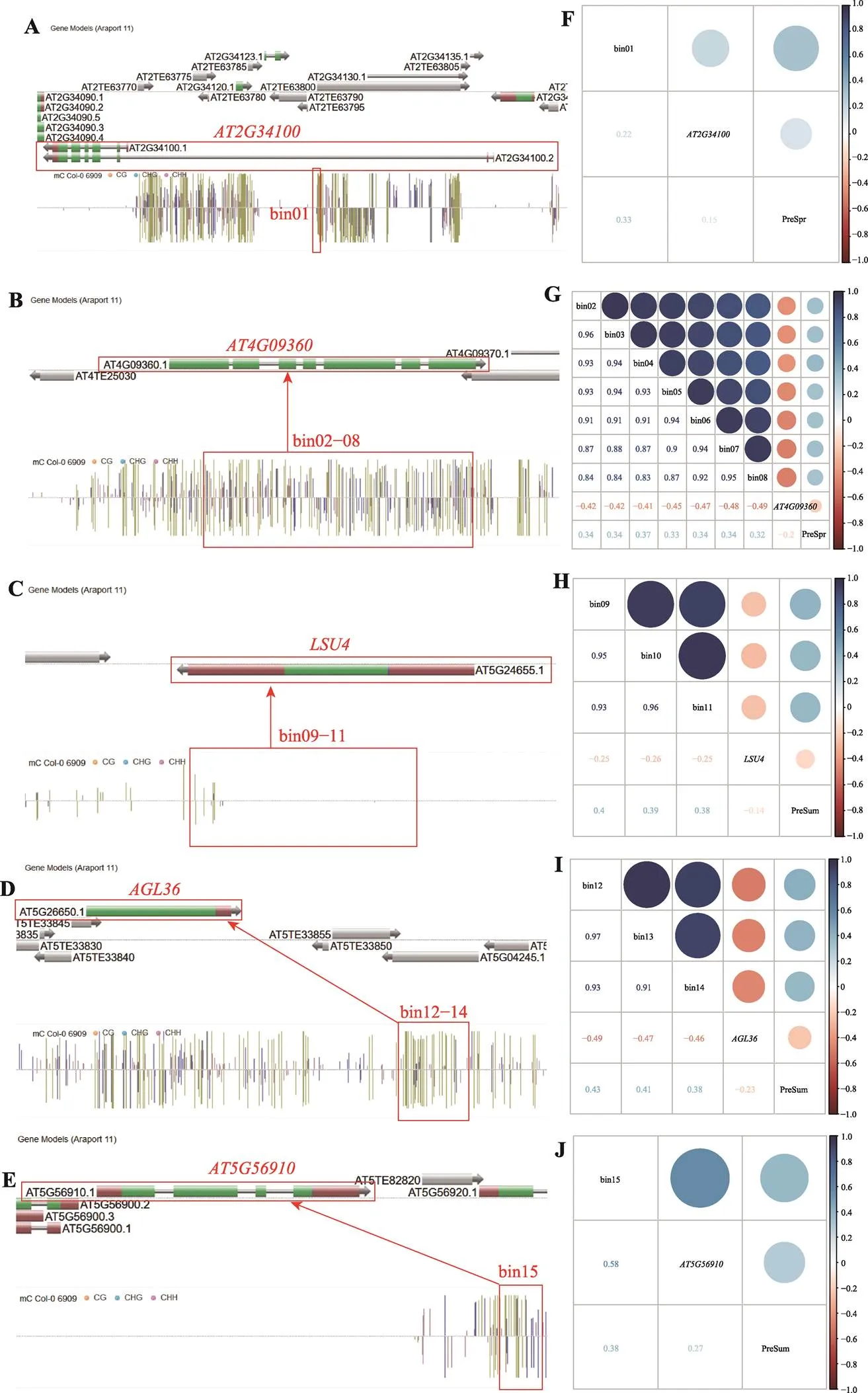
图3 与降水相关表观等位基因基因组位置及相关分析
A~E:Anno-J浏览器[10]截图,分别展示本研究在拟南芥中鉴定到的与降水相关的基因与可能调控它们表达甲基化区域的基因组位置。每小图上半部分展示基因位置,下半部分展示拟南芥Col-0生态型基因组甲基化水平,其中每一个柱形代表一个甲基化位点,颜色、高度和上下方向分别表示甲基化的类型、水平和DNA两条链。F~J:甲基化水平、基因表达量、降水量相关系数。分别以数字和图形两种方式展示(圆圈大小展示关联系数绝对值,从蓝到红对应关联系数1到–1),bin之间高的甲基化正相关性表明它们可能作为一个整体起作用。PreSpr:春季降水量;PreSum:夏季降水量。
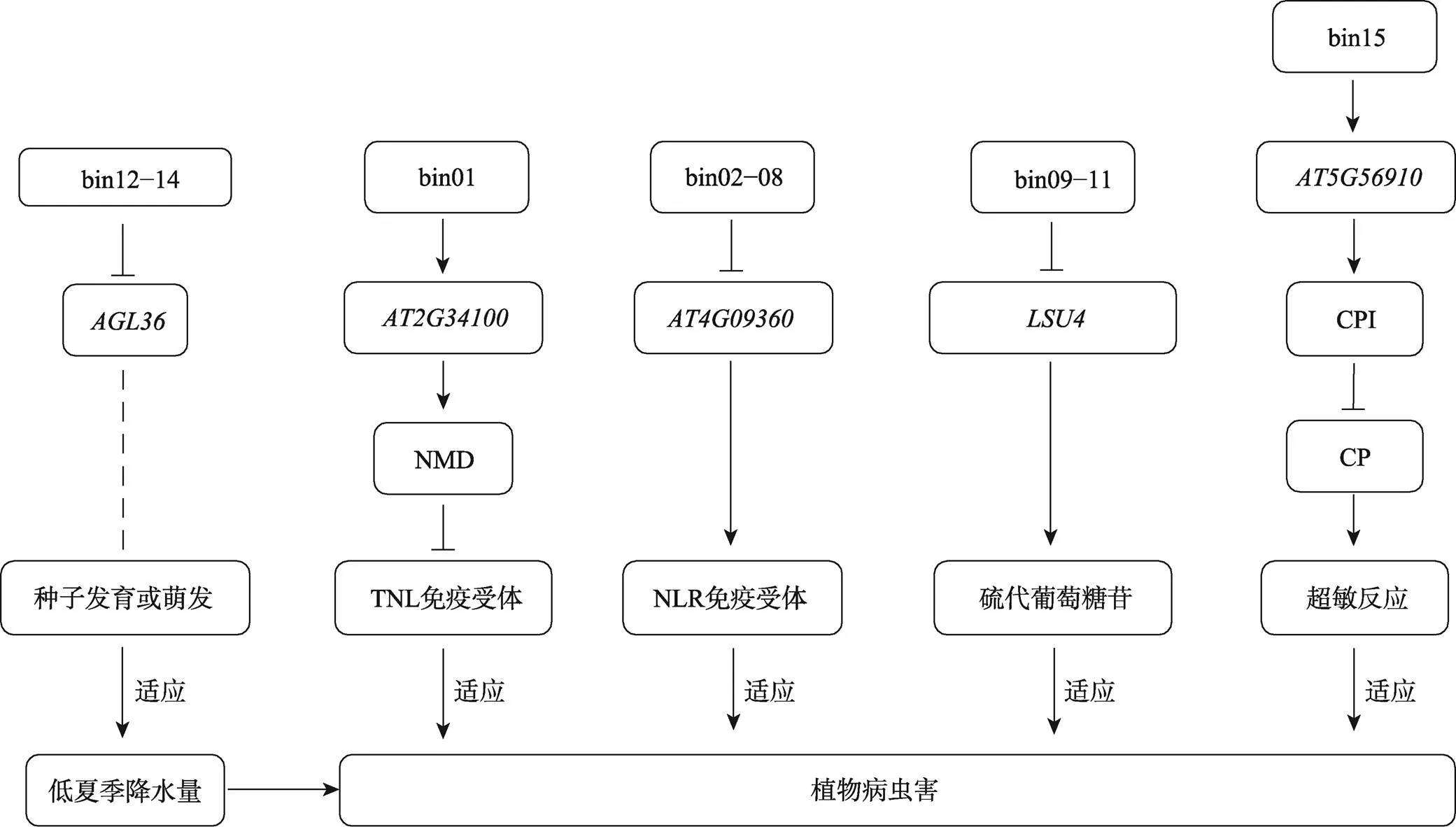
图4 DNA甲基化通过影响基因表达参与拟南芥降水量适应性进化
bin表示此区域的甲基化。对种子发育可能有影响,但具体模式和机制仍不清楚,因此用虚线表示。
2.3 蛋白互作网络和基因富集分析
本研究鉴定到5个可能参与拟南芥对当地降水量适应的基因,而它们附近特定区域的差异甲基化能影响它们的表达,产生对应的“表观等位基因”。为进一步挖掘这些表观等位基因影响拟南芥水分适应的具体机制,提取与这些表观等位基因所涉及的bin相关且与春季或夏季降水量也相关的所有基因(基因集),对每一基因集进行蛋白互作网络、GO功能和KEGG通路分析。结果表明,只有同时与bin09–11和夏季降水量关联的基因集形成了明显的蛋白互作网络(图5A),且基因功能和代谢通路具有显著富集。
对同时与bin09–11、夏季降水量相关的基因集进行GO富集分析,结果表明,“生物学过程”显著富集于硫化合物特别是硫代葡萄糖苷(glucosinolate)代谢途径(图5B),“细胞组分”显著富集于高尔基体和内膜系统(图5C)。KEGG富集分析结果进一步表明这些基因只显著富集于硫代葡萄糖苷代谢途径(图5,D和E)。硫代葡萄糖苷是十字花科植物中一种富含氮和硫的次生代谢产物,在植物生长发育和免疫防御中具有重要作用。芥子酶与硫代葡萄糖苷共存于不同细胞中,当植物组织受损时芥子酶水解硫代葡萄糖苷产生异硫氰酸盐和腈类化合物,前者对多数病原菌和昆虫具有毒害作用[23]。此外,硫代葡萄糖苷及其水解产物能参与植物硫代谢平衡,缓解硫胁迫[24]。在蛋白互作网络中,LSU4蛋白通过直接与APR1、KCS13两个蛋白互作参与整个蛋白互作网络(图5A)。是植物LSU基因家族成员,该基因家族主要与拟南芥低硫胁迫响应相关,但目前还没有研究报道在植物硫代谢途径中的作用[21]。基于以上结果,本研究推测可能也参与拟南芥硫代谢网络,并能通过影响硫代葡萄糖苷的代谢参与拟南芥对生物胁迫的响应。bin09–11的甲基化可能通过抑制的表达降低拟南芥对生物胁迫的抵御能力,而低降水量可能促进当地病虫害的发生,因此在当地夏季低降水量的气候条件下具有bin09–11高甲基化的植株在进化中逐渐被淘汰(图4)。
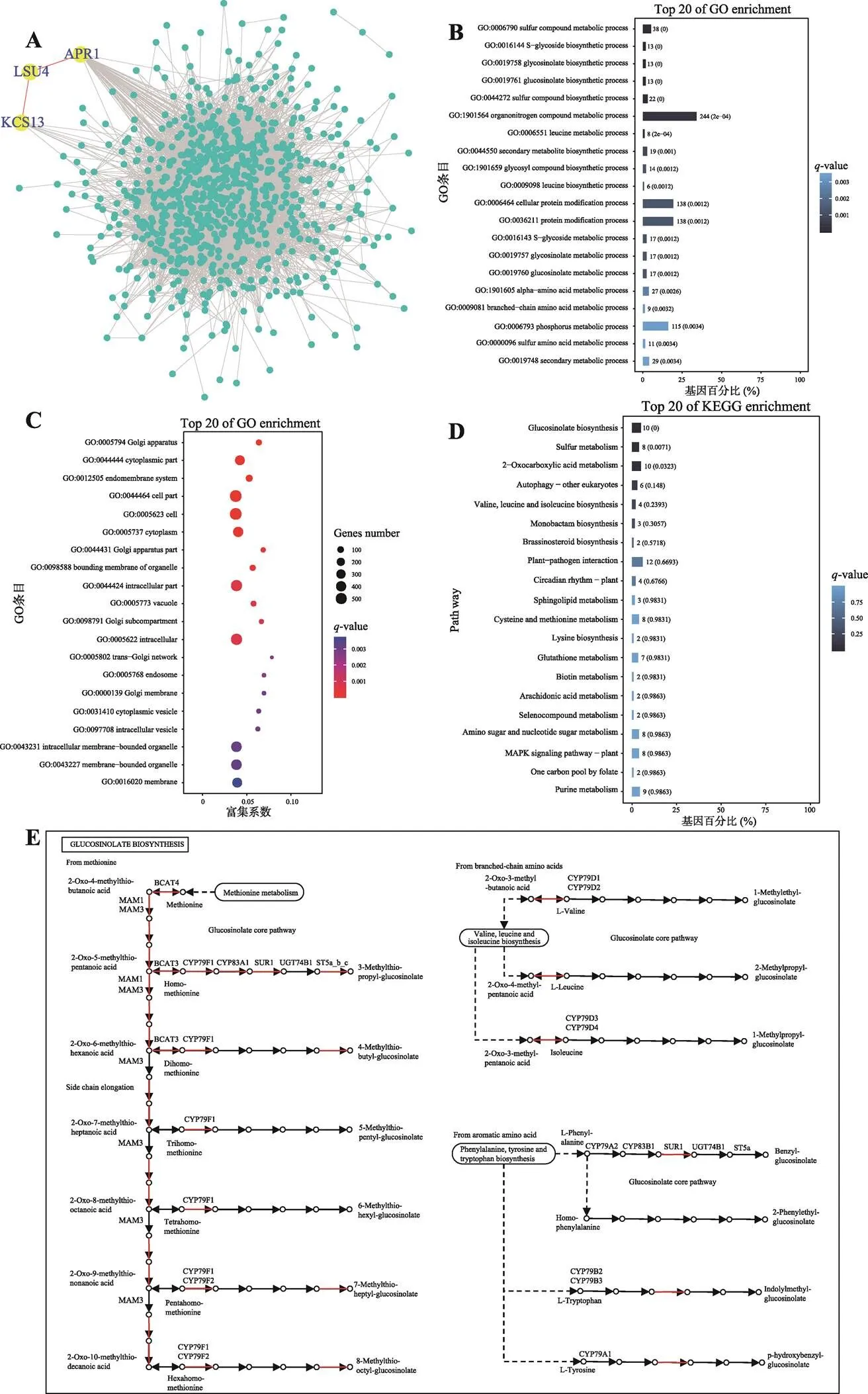
图5 与bin09–11和夏季降水量相关基因PPI、GO和KEGG分析
A:蛋白互作网络。LSU4通过直接与APR1和KCS13互作参与整个蛋白互作网络。B:GO“生物学过程”分析。C:GO“细胞组分”分析。D:KEGG通路分析。E:相关基因在硫代葡萄糖苷代谢途径中的分布。红色线条标注该过程相关基因存在于本研究的基因集中。
3 讨论
从植物自然种群中鉴定表观等位基因并进行功能分析是植物生态表观遗传学研究的重要方面,本研究结果为探究拟南芥中与降水相关表观等位基因的鉴定和功能分析奠定了基础,该方法也同样适用于其他物种和环境因子。利用“拟南芥1001”项目测序的不同拟南芥生态型的甲基化组、转录组和气候数据进行两两关联分析,能挖掘与特定环境因素适应有关的表观等位基因。同时结合蛋白互作网络和基因富集分析,能进一步分析从特定甲基化模式到环境适应的具体机制。参考目前拟南芥和其他物种中已鉴定到的表观等位基因,对基因表达有调控作用的甲基化区域一般位于基因内部或附近,因此在进行关联分析时应着重考虑位于基因内部和附近的甲基化区域。需要特别指出的是,表观等位基因一般是指仅由于DNA甲基化差异引起表达量不同的等位基因,因此在鉴定过程中需排除DNA序列变异的影响。虽然从理论上来说DNA序列变异也会影响甲基化水平,但事实上即便基因组区域存在遗传变异,整体甲基化水平也可能不受影响。如何力等[9]鉴定到控制基因表达的NMR19区域甲基化是独立于该区域遗传变异的。由此可见,遗传因素对表观等位基因的影响是复杂的,单纯依赖生物信息学方法很难分析遗传变异对表观等位基因的影响,本研究鉴定到的表观等位基因仍需设计相关分子遗传学实验进一步验证。此外甲基化与基因表达、基因表达与环境适应(表型)之间的因果性和充分性也需要设计进一步的实验验证。即便如此,本研究的方法和结果仍能为表观等位基因的进一步实验验证和功能分析提供参考,特别是能与基因组定向甲基化编辑技术结合,加快表观等位基因的筛选工作[25]。
本研究以降水量为例鉴定到5个可能参与拟南芥降水量适应性进化的表观等位基因(表1,图4)。这5个基因中,和个基因均直接参与拟南芥免疫反应。其中bin01甲基化对的表达可能具有促进作用,该基因参与拟南芥NMD途径,而NMD途径被证明对拟南芥免疫反应有调节作用。bin02–08的甲基化对可能有抑制作用,该基因是拟南芥免疫受体基因家族成员。编码的蛋白是半胱氨酸蛋白酶抑制剂,与植物局部超敏反应有关,bin15甲基化对该基因表达可能具有促进作用。bin09–11甲基化对可能具有抑制作用,通过蛋白互作网络、GO和KEGG富集分析,本研究发现可能通过影响硫代葡萄糖苷代谢途径参与拟南芥对病虫害的防御。降水量是影响当地病虫害严重程度的重要因素之一,因此本研究推测当地不同的病虫害发生水平可能是对以上、、和这4个基因表达量产生选择压力的直接原因。是受表观遗传因素调控的印记基因,与拟南芥种子发育有关。本研究首次发现可能存在表观等位基因,基因下游1400 bp左右区域的bin12–14甲基化对该基因表达可能具有抑制作用,而该基因能通过调控种子发育影响拟南芥对不同降水条件的适应。
气候数据的全甲基化组关联分析能够揭示环境条件与基因组甲基化位点之间的关系。本研究结果表明基因组大量位点的DNA甲基化与当地气候特征具有很强的关联性,产生这种关联的原因很大程度上是由于这些甲基化位点能控制某些功能基因的表达,而这些基因能影响植物对不同环境条件的适应能力。表观遗传突变在拟南芥自然种群中能产生丰富的表观遗传多样性[26],有利于个体环境适应的突变受到正向选择,这可能是表观等位基因参与植物环境适应性进化的潜在机制。此外,表观等位基因可能介导了植物的拉马克进化,即环境条件直接诱导相关等位基因的产生,使植物对环境产生快 速适应[27],但其中的具体机制还有待进一步证实和解析。
[1] Richards CL, Alonso C, Becker C, Bossdorf O, Bucher E, Colomé-Tatché M, Durka W, Engelhardt J, Gaspar B, Gogol-Döring A, Grosse I, van Gurp TP, Heer K, Kronholm I, Lampei C, Latzel V, Mirouze M, Opgenoorth L, Paun O, Prohaska SJ, Rensing SA, Stadler PF, Trucchi E, Ullrich K, Verhoeven KJF. Ecological plant epigenetics: evidence from model and non-model species, and the way forward., 2017, 20(12): 1576–1590.
[2] Zhang HM, Lang ZB, Zhu JK. Dynamics and function of DNA methylation in plants., 2018, 19(8): 489–506.
[3] Zhao Q, Wang W, Sun YQ. Review on the correlation between DNA methylation and gene expression in plant., 2016, 32(4): 16–23.赵倩, 王巍, 孙野青. 植物DNA甲基化与基因表达的关联性研究进展. 生物技术通报, 2016, 32(4): 16–23.
[4] Kakutani T. Epi-alleles in plants: inheritance of epigenetic information over generations., 2002, 43(10): 1106–1111.
[5] Luan X, Liu SC, Ke SW, Dai H, Xie XM, Hsieh TF, Zhang XQ. Epigenetic modification of ESP, encoding a putative long noncoding RNA, affects panicle architecture in rice., 2019, 12(1): 20.
[6] Ma CQ, Jing CJ, Chang B, Yan JY, Liang BW, Liu L, Yang YZ, Zhao ZY. The effect of promoter methylation on MdMYB1 expression determines the level of anthocyanin accumulation in skins of two non-red apple cultivars., 2018, 18(1): 108.
[7] Song QX, Zhang TZ, Stelly DM, Chen ZJ. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons., 2017, 18(1): 99.
[8] Wei XJ, Song XW, Wei LY, Tang SQ, Sun J, Hu PS, Cao XF. An epiallele of rice AK1 affects photosynthetic capacity., 2017, 59(3): 158–163.
[9] He L, Wu WW, Zinta G, Yang L, Wang D, Liu RY, Zhang HM, Zheng ZM, Huang H, Zhang QZ, Zhu JK. A naturally occurring epiallele associates with leaf senescence and local climate adaptation inaccessions., 2018, 9(1): 460.
[10] Kawakatsu T, Huang SSC, Jupe F, Sasaki E, Schmitz RJ, Urich MA, Castanon R, Nery JR, Barragan C, He YP, Chen HM, Dubin M, Lee CR, Wang CM, Bemm F, Becker C, O'Neil R, O'Malley RC, Quarless DX, Schork NJ, Weigel D, Nordborg M, Ecker JR. Epigenomic diversity in a global collection ofAccessions., 2016, 166(2): 492–505.
[11] Shirzadi R, Andersen ED, Bjerkan KN, Gloeckle BM, Heese M, Ungru A, Winge P, Koncz C, Aalen RB, Schnittger A, Grini PE. Genome-wide transcript profiling of endosperm without paternal contribution identifies parent-of-origin-dependent regulation of AGAMOUS- LIKE36., 2011, 7(2): e1001303.
[12] Walia H, Josefsson C, Dilkes B, Kirkbride R, Harada J, Comai L. Dosage-dependent deregulation of an AGAMOUS- LIKE gene cluster contributes to interspecific incompatibility., 2009, 19(13): 1128–1132.
[13] Ferrero-Serrano Á, Assmann SM. Phenotypic and genome- wide association with the local environment of., 2019, 3(2): 274–285.
[14] Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations., 2012, 28(10): 1353–1358.
[15] Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks., 2003, 13(11): 2498–2504.
[16] Xu FH, Xu BQ. Nonsense-mediated mRNA decay in eukaryotic cells., 2008, 30(1): 55–58.徐飞虎, 许宝青. 真核细胞中无义介导的mRNA降解. 细胞生物学杂志, 2008, 30(1): 55–58.
[17] Wachter A, Hartmann L. NMD: nonsense-mediated defense., 2014, 16(3): 273–275.
[18] Huo ZG, Li MS, Wang L, Xiao JJ, Huang DP, Wang CY. Impacts of precipitation variations on crop diseases and pests in China., 2012, 45(10): 1935–1945.霍治国, 李茂松, 王丽, 肖晶晶, 黄大鹏, 王春艳. 降水变化对中国农作物病虫害的影响. 中国农业科学, 2012, 45(10): 1935–1945.
[19] Zhang L, Huo ZG, Wang L, Jiang YY. Effects of climate change on the occurrence of crop insect pests in China,, 2012, 31(06): 1499–1507.张蕾, 霍治国, 王丽, 姜玉英. 气候变化对中国农作物虫害发生的影响. 生态学杂志, 2012, 31(06): 1499– 1507.
[20] Xia ST, Li X. Open a door of defenses: plant resistosome., 2019, 54(03): 288–292.夏石头, 李昕. 开启防御之门: 植物抗病小体. 植物学报, 2019, 54(03): 288–292.
[21] Sirko A, Wawrzyńska A, Rodríguez MC,Sęktas P. The family of LSU-like proteins., 2015, 5: 774.
[22] Zhang JL, Fu C. Response of cysteine protease and cysteine protease inhibitor to adverse stress., 2018, 16(13): 4453–4459.张洁琳, 付畅. 半胱氨酸蛋白酶及半胱氨酸蛋白酶抑制剂对逆境胁迫的应答. 分子植物育种, 2018, 16(13): 4453–4459.
[23] Yan MJ, Song PL, Huangfu HY, Hao LF, Guo C, Huangfu JR, Jia XQ, Wu J, Shi ZD, Li ZQ. Research progress on the regulation and synthesis genes of the glucosinolate biosynthesis pathway incea., 2019, 47(1): 33–41.燕孟娇, 宋培玲, 皇甫海燕, 郝丽芬, 郭晨, 皇甫九茹, 贾晓清, 吴晶, 史志丹, 李子钦. 芥菜型油菜硫代葡萄糖苷基因的研究进展. 北方农业学报, 2019, 47(1): 33–41.
[24] Sánchez-Pujante PJ, Borja-Martínez M, Pedreño MÁ, Almagro L. Biosynthesis and bioactivity of glucosinolates and their production in plantcultures., 2017, 246(1): 19–32.
[25] Gallego-Bartolomé J, Gardiner J, Liu W, Papikian A, Ghoshal B, Kuo HY, Zhao JM, Segal DJ, Jacobsen SE. Targeted DNA demethylation of thegenome using the human TET1 catalytic domain., 2018, 115(9): E2125–E2134.
[26] Johannes F, Schmitz RJ. Spontaneous epimutations in plants., 2019, 221(3): 1253–1259.
[27] Verhoeven KJ, von Holdt BM, Sork VL. Epigenetics in ecology and evolution: what we know and what we need to know., 2016, 25(8): 1631–1638.
Multi-omics association analysis revealed the role and mechanism of epialleles in environmental adaptive evolution of
Zhichao Mei, Zhujun Wei, Jiahui Yu, Fengdan Ji, Linan Xie
Epialleles, generally referring to alleles whose expression is altered due to differential DNA methylation levels, have important roles in plant morphology, development, and various physiological processes. However, the influence of environmental factors on the plant epialleles under natural conditions is unclear. Meanwhile, the role and mechanism of epialleles in the environmental adaptive evolution of plants remain elusive. In this study, we collected the transcriptome, methylome and climate data from 623accessions, derived from worldwide distributions. Then the data were subject to multi-omics association analysis combined with protein interaction network and gene enrichment analysis to identify epialleles related to specific environmental factors and to explore their possible mechanisms involved in the environmental adaptive evolution of. We focused on spring and summer precipitation and identified five potential epialleles with differential DNA methylation levels located in specific regions of the genes:, AT2G34100, AT4G09360,and AT5G56910. Interestingly, the imprinted generelated to seed development was discovered as an epiallele involved in the environmental adaptive evolution of, and the other four genes are related to the response to biotic stress. By protein interaction, GO enrichment, and KEGG pathway analysis, we also found thatmay participate in the sulfur metabolism network like other members of theLSU gene family, and be involved in the biotic stress response by affecting glucosinolate metabolism. In natural conditions low precipitation may affect the severity of local pests and diseases. Therefore, we speculate that DNA methylation associates with the expression of the four genes to regulate the resistance ofto local pests and diseases, and ultimately participates in the adaptation to local environments.
plant ecological epigenetics; multi-omics association analysis; epiallele; environmental adaptive evolution;
2019-11-15;
2020-02-29
国家自然科学基金项目(编号:31871220),中央高校基本科研业务费专项资金(编号:2572017DA06)和东北林业大学教育教学改革专项资金项目资助[Supported by the National Natural Science Foundation of China (No. 31871220), the Fundamental Research Funds for the Central Universities (No. 2572017DA06), and Special Funds for Education and Teaching Reform of Northeast Forestry University]
梅志超,本科生,专业方向:生物科学。E-mail: zcmei9901@outlook.com
解莉楠,博士,副教授,研究方向:植物抗逆分子生物学。E-mail: linanxie@nefu.edu.cn
10.16288/j.yczz.19-348
2020/3/14 7:00:00
URI: http://kns.cnki.net/kcms/detail/11.1913.r.20200313.1458.002.html
(责任编委: 刘宝)
猜你喜欢
广东气象(2022年5期)2022-10-26
成都信息工程大学学报(2022年3期)2022-07-21
河北果树(2021年4期)2021-12-02
上海公路(2019年3期)2019-11-25
启蒙(3-7岁)(2019年8期)2019-09-10
福建基础教育研究(2019年10期)2019-05-28
江西农业(2018年23期)2018-02-11
上海农业学报(2017年3期)2017-04-10
红领巾·探索(2015年9期)2015-09-10
天然产物研究与开发(2014年6期)2014-04-27
