
单细胞测序技术及其在乳腺癌研究中的应用
2020-04-15 03:15张强顾明亮
遗传 2020年3期
张强,顾明亮
综 述
单细胞测序技术及其在乳腺癌研究中的应用
张强,顾明亮
聊城市人民医院,转化医学研究联合实验室,聊城 252000
乳腺癌是起源于乳腺各级导管和乳腺上皮细胞,由增生到不典型增生而逐步发展成原位癌、早期浸润癌至浸润性癌的一种恶性肿瘤。传统高通量测序技术对乳腺癌的研究主要是鉴定与乳腺癌发生发展相关的“驱动基因”,但是对于乳腺癌基因组结构变化以及亚克隆的鉴定等存在一定的局限性,并且忽略了乳腺癌肿瘤细胞之间的异质性。近年来兴起的单细胞测序技术是以单个细胞为研究对象,对基因拷贝和基因表达等数据进行分析,探究乳腺癌的细胞组成、细胞状态和细胞命运,绘制乳腺癌生态系统图谱,对临床患者进行精准分层,为实现个体化治疗提供支持。同时,还可以揭示乳腺癌与T细胞、巨噬细胞等免疫细胞间的相关性,为发现乳腺癌新的治疗靶点、免疫检查点等提供参考。本文对单细胞测序技术及其在乳腺癌研究中的应用和研究进展进行了综述,以期为单细胞测序技术的进一步发展提供参考,同时为理解乳腺癌的发病机制和免疫治疗提供基础支持。
单细胞测序;乳腺癌;肿瘤微环境;免疫治疗
乳腺癌是乳腺组织中各级导管和乳腺上皮细胞恶性增生的一种癌症,其临床表现为乳房肿块、乳腺形状改变、皮肤上的酒窝、乳头溢出液或出现红色或有鳞的皮肤。乳腺癌分为原位癌和浸润性癌,一般原位癌并不致命,但由于癌细胞间的粘连性降低,癌细胞一旦脱落,会随血液或淋巴液扩散至全身形成转移,引发浸润性癌。据全球癌症监测台(global cancer observatory, GLOBOCAN)的数据显示,乳腺癌高居中国女性癌症发病率首位,严重威胁女性健康[1]。目前的研究表明,环境、遗传与生活方式和乳腺癌发生发展密切相关,营养干预、减轻体重已被证实是有效的一级预防措施[2]。近年来大规模的研究已较为系统地描绘了乳腺癌的致癌因素并勾勒出基因相互作用的网络,揭示了乳腺癌的生物学复杂性,扩大了病患早期诊断、分级治疗和生物标志物靶向治疗的途径,为乳腺癌的精准治疗提供了理论支持和潜在的靶点[3]。针对乳腺癌的靶向治疗,能有效阻断肿瘤细胞之间的信息传递,抑制肿瘤的生长,达到治疗的目的。然而,目前对乳腺癌的靶向治疗导致其产生耐药性已成为临床治疗中的普遍现象,因此亟需发现新的治疗模式以降低乳腺癌的耐药性和复发风险。
传统高通量测序技术主要被用于新癌症基因的发现和证明瘤内的异质性。乳腺癌的全基因组测序表明,体细胞突变类型中的“驱动”突变(driver mutations)会促进肿瘤的发展,而“搭车”突变(passenger mutations)可能是基因组不稳定的产物,但值得注意的是“驱动”突变和“搭车”突变之间在乳腺癌发展中会发生逆转[4,5]。Stephens等[6]发现在测序的100种乳腺癌中,有73种不同的癌症基因突变组合,其中的6种组合可以归纳到JUN激酶途径中。Shah等[7]在104例三阴性乳腺癌(triple-negative breast cancer, TNBC)病例中发现,确诊时TNBC表现出广泛而连续的基因组进化谱,并且大多数都包含数百个体细胞突变。通过对其进行基因组和转录组测序,发现常见的乳腺癌突变类型多数与p53信号通路、磷脂酰肌醇激酶PIK3和表皮生长因子受体酪氨酸激酶ERBB信号通路有关。虽然乳腺癌突变基因和突变组合存在多样性,但与其相关的信号通路却具有高度一致性。正是由于信号通路存在一致性,其乳腺癌表型可能相似。由于细胞间的异质性,即使相同表型的细胞也存在遗传信息的差异,很多分子信息会在整体分析中丢失。这意味着通过传统的高通量测序对乳腺癌进行更精确的分子分型会更加的困难。为了弥补传统测序技术的局限性,单细胞测序技术则应运而生。
单细胞测序技术是以单个细胞为起始材料,允许在单细胞水平上进行拷贝数和基因表达的分析,从而通过构建以非线性分支结构为特征的系统发育树,绘制癌细胞多样性和克隆进化的清晰图像[8~11]。癌症作为一种基因组疾病,从原发性肿瘤,经过循环肿瘤细胞,到转移性肿瘤,涉及到一系列基因组的变化[12~14]。在细胞水平上,癌细胞的异质性早已被染色体核型分析[5]和组织切片荧光原位杂交(fluorescence in situ hybridization, FISH)所证实[15]。乳腺癌常常表现出瘤内基因组的显著异质性,其克隆的多样性影响了临床的诊断和治疗[16]。单细胞测序技术的应用,为了解乳腺癌生态系统的发生和进化,以及免疫治疗机制提供了可能[17]。本文对单细胞测序的技术进行了介绍,并对其在乳腺癌研究中的应用和进展进行了综述,为绘制乳腺癌基因突变图谱和揭示其免疫治疗的机制提供参考。
1 单细胞测序技术
传统高通量测序技术需要从大量的细胞中获取足够的DNA样本,因此测序数据是这些细胞整体的特征信息。而单细胞测序技术是在单个细胞水平上,对基因组、转录组和表观遗传组等进行高通量测序分析,能够揭示单个细胞的基因结构和基因表达动态,反映细胞间的异质性。单细胞测序的技术流程包括单细胞捕获、基因组/转录组/表观组的文库构建、测序与数据分析等步骤(图1)。其中,单细胞捕获和文库构建的质量是评价是否能顺利完成测序的关键。
1.1 单细胞制备
1.1.1 单细胞悬液准备
制备高质量的单细胞悬浮液是单细胞研究成功的关键。不管起始样本是什么,单细胞悬液的状态对于有效的单细胞捕获和后续的单细胞建库测序是至关重要的。对于悬浮的液体样本(如血液样本),可进行密度离心,然后直接用于单细胞的捕获。对于生物组织须首先利用机械和酶处理进行组织解离。组织通过机械切割或刀片切碎,之后利用酶促消化用于分离细胞。常用的酶类型包括胶原蛋白酶、细胞消化液(accutase)、胰蛋白酶,以及商业化的酶混合物,如Liberase T。不同组织所使用的酶和消化时间是不同的[18~32](表1)。在组织裂解过程中会形成细胞团块,可使用DNase I减少细胞团块的形成。此外,对于较敏感的细胞类型可能在样品制备过程中被损坏,因此酶处理时间应保持在所需的最低限度。
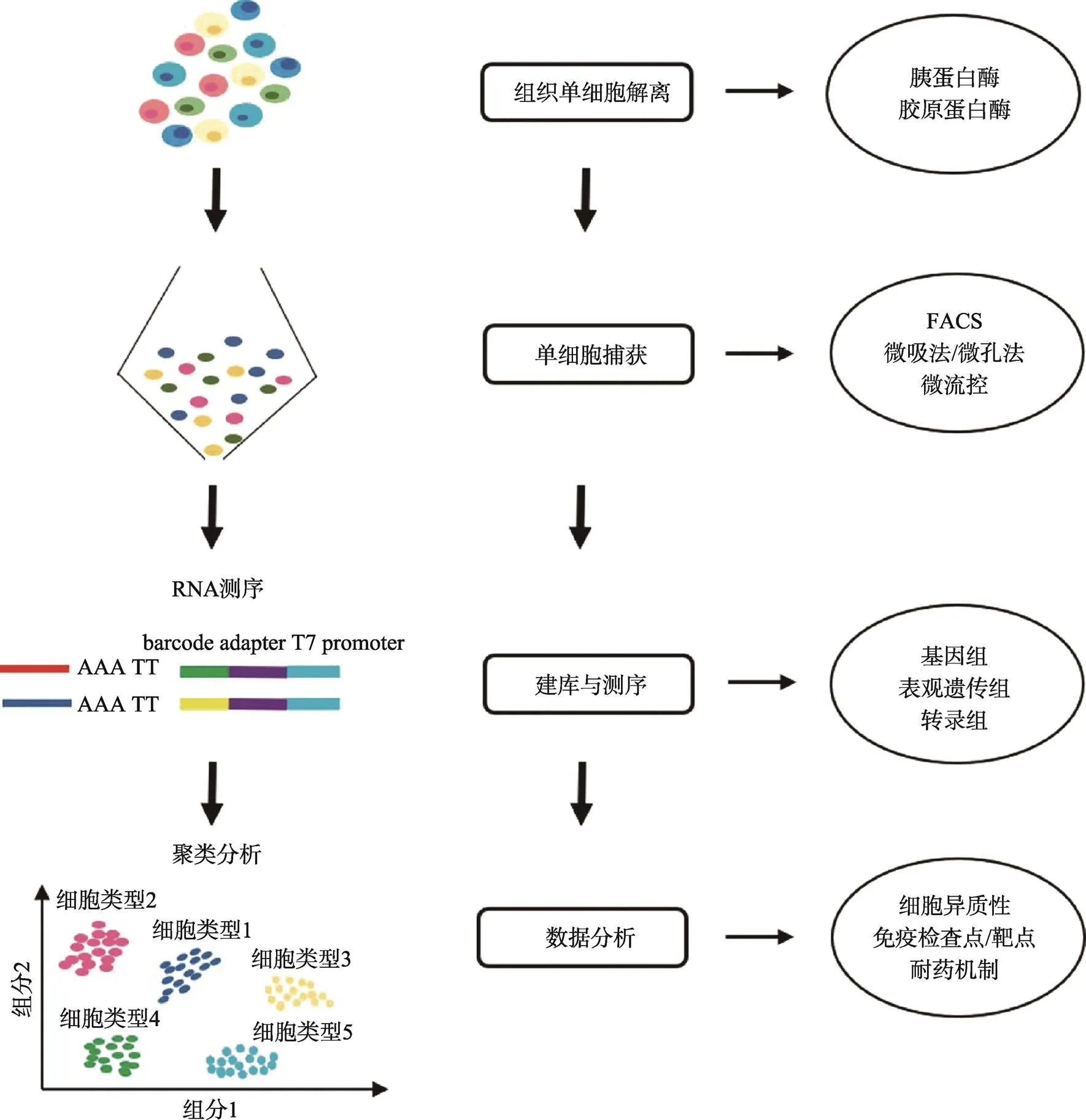
图1 单细胞测序技术的流程图
利用胰蛋白酶和/或胶原蛋白酶等对组织样本酶解消化,获得得单细胞悬液。然后,通过FACS、微流控等对单个细胞捕获。随后对其基因组、转录组或表观遗传组进行建库并测序,通过生物信息学分析解读测序数据。

表1 组织特异性酶处理制备单细胞悬浮液
在单细胞捕获之前,裂解分离的细胞悬液需要洗涤或利用过滤器进行清洗,以除去大团块和碎片。细胞悬液的洗涤,洗涤液成分、离心条件和/或过滤器类型都会影响细胞悬液的纯度和质量。在细胞洗涤时,理想离心条件下,会产生一团固体团块,但该团块的结构不能太过紧实,并且使残留在上清中的细胞最少。对于较大的细胞,如大多数未成熟的细胞系,室温条件下150 r/min可离心3 min,对于较小的细胞,如外周血单核细胞(peripheral blood mononuclear cell, PBMC),室温条件下300 r/min离心需5 min[33]。利用过滤器过滤时,其过滤器孔径应该大于样品中细胞的最大直径,但要小到足以捕捉较大的团块。根据凝块的程度和过滤器的类型,细胞的数量和洗涤液的量可以有所不同。一般情况下,推荐使用MACS智能滤器,因为其对细胞浓度的影响很小,对于浓度较低的细胞悬浮液体积,建议使用Flowmi™尖端过滤器,使体积的损失最小化[33]。细胞清洗液和重悬液推荐使用含0.04%牛血清白蛋白的磷酸盐溶液(phosphate buffered saline, PBS),添加牛血清白蛋白使细胞损失和聚集最小化[33]。对于敏感型细胞,如原代细胞、干细胞等,为最大限度提高其生存能力,可用细胞培养常用缓冲液Hank’s平衡盐溶液(Hank’s balanced salt solution, HBSS)代替PBS。如果在这些缓冲液中细胞不能维持良好的活力,也可在最常见的细胞培养基中清洗和重悬,如DMEM培养基和M199培养基[33]。洗涤和重悬细胞时,其浓度要维持在低于5000细胞/µL,较高浓度的细胞会导致细胞聚集成团,从而干扰理想的单细胞悬浮液的生成。为尽量减少剪切力对细胞的物理损伤,在细胞洗涤和重悬过程中,建议使用宽口径的移液管,因为常规移液管的尖端,容易对细胞造成剪切和破坏。细胞悬浮液应在制备后,30 min内应尽快进行后续实验,以保证细胞悬浮液质量。
1.1.2 单细胞捕获
单细胞测序的第一步且最关键的一步就是如何有效的捕获单个活性细胞。单细胞捕获的最大挑战是保证细胞的活性和完整性[34]。当前组织酶解单细胞获取方法有微吸法[35]、微孔法[36]、流式细胞分选(fluorescence-activated cell sorting, FACS)和微流控(microfluidics)。对于组织切片常用的单细胞获取方法有激光捕获显微切割(laser capture microdissection, LCM)和膜片钳(patch clamping)。
微吸法和微孔法主要是依靠手工操作来获取单个细胞。微吸法是在显微镜视野下,用微量移液器或口吸管吸取单细胞,其获取的细胞数量较少,并且需要具备很强的实验操作技能。这两种方法是可以直接观察到吸取的细胞,依据细胞的形态能够确保细胞的完整性,从根本上保证了细胞的质量。
FACS是把制备好的单细胞悬液通过流式细胞仪,根据细胞的特异性分子标记、粒径、颗粒密度等参数,将单个所需细胞依次打入每个孔中[37]。细胞通量大和分离速度快是FACS的主要优势。由于FACS需要大量的单细胞悬液作为起始材料,所以它较难从数量较少的细胞群中分离得到单个细胞,如循环肿瘤细胞[38]。
微流控是当前获取单个细胞最常用的方法,它是通过控制微流体芯片中的液体流动来捕获单个细胞。目前应用最广泛的微流控系统是FluidigmC1系统和10×Genomics Chromium系统。Fluidigm C1芯片上的细胞捕获位点有800个,不仅能满足实验要求,并且后续的反转录和cDNA扩增可以自动化地完成[39]。商业化的10×Genomics是利用油相的微小水滴封装单细胞,被捕获的单细胞在这样的油包水液滴中裂解,并被标记,之后再进行反转录和扩增[40,41]。
LCM和膜片钳是从组织切片上获取单个细胞,因此可以获知细胞在组织中的空间位置[42,43]。LCM 利用激光束切割目的细胞,由于细胞之间的粘连性,易受到边缘细胞的污染[44]。这两种方法获取的细胞尽管通量低,但在细胞形态学或染色体特性的视觉检查上具有明显的优势。
1.2 单细胞建库与测序技术
1.2.1 单细胞全基因组建库与测序
单细胞全基因组测序是对单个细胞中的微量全基因组DNA进行扩增,在获得高覆盖率的基因组后进行高通量测序,以揭示细胞间差异和细胞的进化(表2)。
单细胞的基因组学研究中,由于细胞内 DNA 的含量极少,因此首先需要通过全基因组扩增技术将 DNA 进行扩增[45]。全基因组扩增(whole genome amplification, WGA)是一种对极低起始量的基因组进行非选择性扩增的技术,是在没有序列倾向性的前提下大幅度增加DNA总量,以满足后续分析需求[46]。连接锚定PCR(ligation anchored PCR, LA- PCR)是先进行DNA的剪切或消化,然后进行接头的连接,之后再进行PCR扩增[47]。引物延伸预扩增PCR (primer extension pre-amplification PCR, PEP- PCR)是在热循环条件下,使用DNA聚合酶进行多轮随机引物扩增,产生多个基因组DNA序列[48]。简并寡核苷酸引物PCR (degenerate oligonucleotide- primed PCR, DOP-PCR)是采用具有简并碱基的杂交寡核苷酸引物,在DNA模板上聚集,分两步PCR进行,第一个阶段促进引物在模板上的延伸,第二个阶段进行扩增子的复制[49,50]。这3种基于PCR的单细胞基因组扩增方法具有较高的扩增偏倚,易导致其覆盖率极低且不均匀[51]。

表2 单细胞全基因组建库与测序方法
质粒和病毒的滚环复制机制为多重置换扩增法(multiple displacement amplification, MDA)提供了灵感[52]。MDA是将随机引物退火到变性DNA上,然后在φ29 DNA聚合酶催化下进行恒温链置换合成。与基于PCR的方法相比,MDA降低了扩增偏倚,并产生了更大的扩增子,平均长度>12 kb。此外,φ29 DNA聚合酶的校对活性将错误率降低到106~107 [51]。虽然MDA解决了基于PCR扩增带来的偏倚,但是,当对二倍体人类单细胞基因组进行单核苷酸变异(single nucleotide variations, SNVs)基因分型时,MDA可能会无法检测到两个等位基因,导致将杂合位点误称为纯合位点[53]。
多重退火循环放大扩增(multiple annealing and looping-based amplification cycles, MALBAC)是将MDA与PCR相结合,引入了线性预扩增,以减少与非线性扩增相关的偏倚[9]。引物由随机序列和通用标记序列所组成,在等温链置换反应中,退火到DNA模板上,由嗜热芽孢杆菌DNA聚合酶(DNA聚合酶)进行延伸扩增。在准线性放大阶段进行了五个周期的退火、延伸和变性,以限制反应速率。新合成扩增子的末端与每个引物的序列相同,可以形成闭环,防止它们再次被复制。MALBAC的扩增偏倚低,覆盖率高。但不能对复杂的DNA二级结构进行有效扩增。
在单细胞全基因组建库及测序中,基因组扩增是其关键步骤,可根据研究目的选择适宜的扩增技术或技术组合。DNA以半保留方式进行高保真复制,尽管DNA聚合酶具有校对活性,但碱基对错配是不可避免的。在未来,发现并采用更高保真度的DNA聚合酶,而不是DNA聚合酶、φ29 DNA聚合酶和DNA聚合酶,以降低扩增错误率是单细胞全基因组建库与测序技术优化的方向。
1.2.2 单细胞转录组建库与测序
单细胞转录组测序首先需要将单个细胞中的mRNA进行反转录成cDNA,之后对cDNA进行PCR扩增,在获得足够的基因组后进行高通量测序。如表3所示,现有的单细胞转录组文库构建和测序原理有3种:添加ployA尾巴、基于5′模板置换和体外转录扩增。
传统的mRNA测序方法是对一群细胞进行转录分析,其数据为细胞的平均表达模式。然而,即使是同一个细胞系,单个细胞间基因表达和蛋白质表达水平也不尽相同[54,55]。Tang等[56,57]率先将单细胞RNA技术与二代测序相结合,使单细胞转录组的单碱基分辨率和高通量分析成为可能。在Tang等的方法中,单细胞被直接裂解,释放出来的mRNA被带有锚定序列UP1的寡核苷酸-dT引物反向转录成cDNA。末端脱氧核苷酸转移酶将polyA尾加到cDNA第一链的3′端。利用另一个锚定序列UP2的寡核苷酸- dT引物合成cDNA第二链,利用UP1引物和UP2引物进行PCR扩增,扩增所得到的cDNA足以构建测序所需的文库。Quartz-seq[58]与Tang等的方法类似,唯一不同的就是cDNA第二链合成时使用的依然是和第一链合成时一样的锚定序列UP1的寡核苷酸-dT引物,扩增过程中的副产物会形成发卡结构而不能继续扩增,从而提高了保真度。Quartz-seq和其他polyA加尾的转录组扩增方法一样,具有3′偏向性。Quartz-seq进行PE50 (双端50 bp)测序,平均每个细胞的测序数据量约为1~20×106Reads。SUPeR-seq (single-cell universal polyA-independent RNA-seq)利用含有固定锚定序列的随机引物合成cDNA第一链,引物含有随机核苷酸和寡核苷酸-dT,对多聚腺苷酸和非多聚腺苷酸RNA进行测序[59]。SUPeR-seq进行PE100 (双端100 bp)测序,平均每个细胞的测序数据量约为10,911 Reads。MATQ-seq (multiple annealing and dc- tailing based quantitative RNA-seq)是利用含有G、A和T碱基的引物先进行10个周期的退火,该引物是基于MALBAC引物的改进,反转录后对第一链cDNAs进行Ploy C加尾,再使用G富集的引物高效地合成第二链,UMI(unique molecule identifier)是在第二链合成时引入,在PCR扩增前进行cDNA的特异性标记,其基因组覆盖率高,且非常灵敏[60],MATQ-seq平均每个细胞的测序数据量约为1×106Reads。

表3 单细胞转录组建库与测序方法
SMART-seq (switching mechanism at the 5′end of the RNA template)使用莫洛尼小鼠白血病病毒(moloney murine leukemia virus, M-MLV)反转录酶来完成反转录过程[61~63]。这种酶可以在cDNA第一条链的3′末端加上2~6个胞嘧啶(C)。该方法倾向于在5′端完成模板转换,所以这是一种扩增全长转录本的方法。SMART-seq2是在SMART-seq的基础上,把模板转换寡核苷酸(template switching oligos, TSO)的最后一个碱基G做了一个锁相核酸(Locked nucleic acid, LNA)修饰,提高了cDNA的产量和敏感性[64,65]。SMART-seq/SMART-seq2,进行PE50 (双端50 bp) 测序,平均每个细胞的测序数据量为1~20×106Reads。Drop-seq是一种能够快速分析成千上万个单细胞的方法。通过将每个细胞与含有barcode和UMI的微珠共包裹在纳米级液滴中,细胞在这个液滴中完成细胞裂解、RNA 捕获和反转录,之后再进行文库扩增以满足后续的测序要求[66]。Drop-seq进行PE50 (双端50 bp)测序,平均每个细胞的测序数据量为737,240 Reads。STRT-seq (single-cell tagged reverse- transcription)是利用寡核苷酸-dT 引物对单个细胞中的mRNA进行反转录[67]。由于反应混合物中含有由鸟嘌呤(G)和barcode组成的寡聚物,cDNA合成时,5′端被M-MLV逆转录酶标记。多个样本的cDNA混合,用单引物进行PCR扩增,扩增后的cDNA纯化、片段化,并准备用于测序的文库。STRT-seq是一种基于5′条形码(barcode)的特异的单细胞转录组测序方法,显示出强烈的5′偏倚。STRT-seq平均每个细胞的测序数据量约为250,000 Reads。STRT- seq-2i[68]是一个在STRT-seq的基础上开发的具有9600个位点,与限制稀释或FACS相结合的纳米孔微阵列平台,细胞通过限制稀释或直接可寻址的FACS排序进行加载。FACS定位的细胞允许index排序,依据细胞属性(如荧光信号或大小)分配阵列坐标和barcode。含有细胞的纳米孔可以通过靶向分配的方式被专门利用,这大大降低了试剂的成本和环境RNA的污染。为了保证在载入纳米细胞的过程中获得高的细胞存活率,可以在缓冲液中加入氟化碳,样品保存在冰上。STRT-seq-2i平均每个细胞的测序数据量约为62,000 Reads。SCRB-seq[69](single cell RNA barcoding and sequencing)依赖模板转换逆转录酶,利用含有barcode、UMI、接头和寡核苷酸-dT的引物反转录单细胞的mRNA得到cDNA。SCRB-seq使用一种改进的基于转座子的碎片法将来自多个细胞的cDNA汇集、扩增,用于多路重复测序,保留并丰富了3′链信息。SCRB-seq平均每个细胞的测序数据量约为165 000 Reads。
CEL-seq (cell expression by linear amplification and sequencing)利用含有barcode、接头和T7启动子的寡核苷酸-dT的引物反转录单细胞的mRNA 得到cDNA[70]。在cDNA的第二链合成后,将多个样本的cDNA聚合均一化进行体外转录(transcription, IVT),之后再进行RNA片段化和反转录以达到扩增的效果。正是由于在3′端引入barcode进行后续的反应,因此CEL-seq具有严重的3′偏倚[71]。CEL-seq2[72]是在CEL-seq的基础上引入了UMI,同时缩短了Barcode、接头和T7启动子引物的长度,提高了反转录效率。在cDNA合成后,会进行一步在CEL-seq中没有的核酸纯化,纯化完成后,其后续的步骤与CEL-seq相同。同样CEL-seq2也具有较高的3′偏倚。CEL-seq/CEL-seq2平均每个细胞的测序数据量为300 000 Reads。In Drops[73](indexing droplets)与Drop-seq是非常相似的,也是利用液滴微流控技术,将细胞和裂解液、反转录试剂和水凝胶微粒共封存在液滴中,在液滴里面自动完成细胞裂解和cDNA的合成。与Drops不同的是,In Drops所用的水凝胶微粒上还含有T7 RNA聚合酶启动子。In Drops平均每个细胞的测序数据量为10,237~ 208,231Reads。MARS-seq[74](massively parallel RNA single-cell sequencing)是一种自动化的大规模并行RNA单细胞测序框架,通过多重RNA测序对数千个细胞进行活体采样,同时严格控制扩增偏差和标记错误。该方法基于FACS将单个细胞分选到384孔板,并随后进行自动化处理。在384孔板进行细胞的裂解,随后含有barcode、接头和T7启动子的寡核苷酸-dT的引物将mRNA 反转录得到cDNA。多个样本的cDNA均一化后进行体外转录(transcription, IVT),之后再进行RNA片段化和反转录扩增。MARS-seq平均每个细胞的测序数据量约为22,000 Reads。
1.2.3 单细胞表观遗传建库与测序
单细胞基因组和转录组的测序数据证明两者不能完全展示细胞的全貌,因为其功能由表观遗传状态所调控,包括DNA甲基化(5-mc)、羟甲基化(5-hmc),组蛋白修饰、非编码RNA调控、染色质重构以及与染色质结合的结构调节蛋白等。
DNA甲基化在基因表达调控中起着重要作用,其在基因转录起始位点附近的存在与相应基因的表达水平降低有关。基于亚硫酸氢盐转化的高通量DNA甲基化测序方法已被用于单细胞DNA甲基化的全基因组测序。scRRBS[75](single-cell reduced representation bisulfite sequencing)是将原有的高通量亚硫酸氢盐转化DNA甲基化的测序方法应用到单细胞水平上。在scRRBS中,所有过程都集中在同一个反应管中,包括基因组的I酶解消化、末端修复和dA-加尾、接头连接和亚硫酸氢盐的转化。亚硫酸氢盐处理后的DNA通过PCR扩增得到足够的DNA用于文库制备和后续测序。scRRBS进行PE100(双端测序,每端100 bp)测序,其数据量为0.5~1.5×106Reads。scBS-seq[76](single-cell bisulfite sequencing)与scRRBS不同,其先进行亚硫酸氢盐转化,之后用寡核苷酸标记的随机引物对亚硫酸氢盐处理过的DNA进行5个周期的引物延伸。捕获寡核苷酸锚定DNA后,用寡核苷酸标记随机引物再进行扩增,产生扩增子,并对扩增子进行扩增以产生足够的DNA测序文库。scBS-seq进行PE100测序,其数据量为3.7~3.9×106Reads。scRRBS和scBS-seq主要缺陷是亚硫酸盐转化过程中DNA急剧降解和亚硫酸盐转化后DNA的纯化,可能导致高偏倚、低覆盖率。此外,scRRBS和scBS-seq不能区分5-mc和5-hmc。
组蛋白N-末端氨基酸残基可发生乙酰化、甲基化、磷酸化、泛素化等多种共价修饰作用,会影响组蛋白与DNA双链的亲和性,从而改变染色质的疏松或凝集状态,来发挥基因调控作用。scChIP-seq[77](single-cell chromatin immunoprecipitation followed by sequencing)是在单细胞分辨率下对组蛋白翻译后修饰,染色质的开放(H3K4me3)或封闭(H3K27me3)状态进行分析。基于inDrop微流控平台,形成一个含有单个细胞的核小体和携带T7启动子、barcode和接头的水凝胶珠的油包水结构。在液滴中完成核小体的接头连接后,进行蛋白免疫相互作用,用抗体把和染色质相互作用的蛋白沉淀下来,捕捉细胞内动态的、瞬时的蛋白质与DNA之间的相互作用,从而获取与其相结合的DNA序列,进行PCR扩增制备测序文库。scChIP-seq进行PE150(双端测序,每端150 bp)测序,其数据量为1630~10 228 Reads。scATAC-seq[78,79](single-cell assay for transposase- accessible chromatin)是对单细胞中的核小体DNA进行测序,其关键步骤是在细胞裂解之后对DNA 进行的转座(如使用Tn5转座酶),对转座后的DNA,用含有barcode、接头等的引物进行PCR扩增,制备测序文库。scATAC-seq 可以基于微流控或FACS的基础上,完成后续的文库构建。scATAC-seq所产生的数据量约为502~69.847 reads。
无论是单细胞基因组、转录组还是表观组测序,对于组织来说,在制备单细胞悬液的同时,保存每个细胞的位置信息尤为重要,因为组织内的位置是细胞识别的关键决定因素。空间转录组[80](spatial transcriptomics)则允许在单细胞水平上同时研究细胞异质性和空间差异。该方法在组织切片上进行转录的分析,不需要进行细胞解离,将组织切片置于含有barcode的逆转录试剂的载玻片上,试剂对组织进行渗透,最终组织被酶解,cDNA则与载玻片上寡核苷酸结合。然而,该方法对揭示细胞的异质性存在偏差。将空间转录组发展成精准的单细胞方法需要保证单细胞层厚度的均一性,在技术上尚有很大的提升空间。
2 单细胞测序在乳腺癌中的应用
2.1 探究乳腺癌细胞异质性
Navin等[17]在2011年通过单细胞基因组测序对乳腺癌的肿瘤群体结构和演化进行研究,精确地量化单个细胞核内的基因组拷贝数,发现3个不同的克隆亚群可能代表不同克隆的序列扩增。他们对其中一个原发性肿瘤进一步分析发现,一个单克隆扩增形成了原发性肿瘤并撒下了转移的种子。此外,还发现了一个意想不到的丰富的亚群,即基因多样化的“假二倍体”细胞,它们不会迁移到肿瘤部位,与肿瘤进展的渐进模型相反,并证明了乳腺癌肿瘤是以不间断的克隆扩张的形式不断生长的。正是因为乳腺癌的不同克隆亚群的不间断生长,使得乳腺癌分化出不同的亚型。乳腺癌的表达谱分析显示其具有5个亚型,这些都和雌激素(ER)、孕酮(PR)和表皮生长因子(Her2)受体相关,而三阴性乳腺癌(ER–/PR–/Her2–)在这些癌症亚型中突变数量是最多的。单细胞测序分析发现三阴性乳腺癌中存在高水平的体细胞突变、TP53的频繁突变(83%)和复杂的非整倍体重排(80%),使肿瘤细胞的克隆多样性增多,导致了广泛的瘤内细胞异质性[81,82]。Chung等[83]对来自11名乳腺癌患者的515个细胞进行单细胞转录组测序,将乳腺癌细胞与非癌细胞分离。乳腺癌细胞在肿瘤内表现出共同的特征,并在乳腺癌亚型和关键的癌症相关通路上表现出瘤内细胞异质性。非癌细胞多数是免疫细胞,有3种类型的细胞:T淋巴细胞、B淋巴细胞和巨噬细胞。T淋巴细胞、调节型T细胞和巨噬细胞均表现一定的免疫抑制特性,而衰竭型T细胞和M2表型的巨噬细胞表现出促癌作用。这些证据表明乳腺癌广泛的瘤内细胞异质性,是由肿瘤微环境中的肿瘤细胞和免疫细胞共同形成的。单细胞图谱加深了人们对乳腺癌生态系统的理解,有助于实现基于肿瘤微环境的患者分类,实现针对其免疫微环境的个体化治疗。
2.2 探究乳腺癌发生和转移机制
乳腺癌基因组测序发现体细胞突变中,驱动突变赋予了癌细胞克隆选择的优势,并与致癌基因存在因果关系,而乳腺癌的驱动突变和突变过程尚未得到全面的探索。Stephen等[6]对100例乳腺癌患者肿瘤的基因组体细胞拷贝数变化和编码外源性蛋白基因的突变进行分析,发现体细胞突变的数量在个体肿瘤之间存在显著差异,其突变数、癌症诊断年龄和癌症组织学分级之间存在很强的相关性,并观察到多种突变特征,如在10%的乳腺癌中存在一种突变,其特征为在TpC二核苷酸处存在大量胞嘧啶突变。该研究团队分析的100例乳腺癌中,其每例体细胞突变过程中碱基的替换和/或插入的总数有很大变化,突变模式也存在相当大的多样性,至少发现40个与乳腺癌发生相关的癌基因的驱动突变,以及73种不同的突变癌症基因组合。总的来说,突变的癌症基因和不同突变组合对遗传事件起着推动作用,多种驱动因素的存在与肿瘤的克隆进化相关,进而引起乳腺癌的遗传多样性。
乳腺癌中突变的癌症基因和突变过程的全景正变得越来越清晰。通常,少数基因会发生突变,但这些经常发生突变的少数基因共同在无数个不同的组合中发挥作用就会诱发癌变。乳腺癌的突变多样性,促使乳腺癌产生不同的亚型,如乳腺导管原位癌(ductal carcinoma, DCIS),组织病理学中分为肿瘤细胞原位和侵袭性(invasive ductal carcinoma, IDC)两类。DCIS是早期乳腺癌最常见的一种形式,由于导管中肿瘤细胞数量有限[84],基质细胞数量较多[85],关于DCIS侵袭和进化知之甚少[86~88]。Casasent等[89]开发了一种结合激光弹射和单细胞DNA测序的方法—TSCS (topographical single cell sequencing)来测量单个肿瘤细胞种基因组拷贝数的变化,同时保存它们在组织切片上的空间信息。利用TSCS追踪了10例DCIS-IDC患者冷冻肿瘤样本在侵袭期间的克隆进化,结果证实原位和侵袭性肿瘤亚群之间存在直接的基因组谱系,大多数突变和拷贝数畸变在侵袭之前就在导管内进化完成。乳腺癌中多数的体细胞突变,包括TP53和PIK3CA驱动突变,都是在侵袭前,即肿瘤进展的早期阶段,在导管内获得。该研究结果支持多克隆侵袭模型,在该模型中,一个或多个克隆肿瘤细胞相互协同从基底膜中逃逸并迁移到邻近组织中,从而建立侵袭性癌。上述发现与癌细胞入侵模型形成对比,这些模型提出不同的克隆可在原位产生侵袭性肿瘤细胞,并反对外部刺激(即“场效应(field effect)”)可导致多灶性疾病的观点。然而,对于克隆肿瘤的逃脱,是完整基底膜破裂后的随机逃脱,还是肿瘤克隆通过克隆之间相互作用,或共栖(commensalism)发生,即单个“先遣”克隆(a single leader clone)破坏、突破基底膜,为后续“追随者”克隆(follower clones)的逃脱扫清道路,尚需要更进一步的功能研究。
2.3 探究乳腺癌免疫治疗机制
2.3.1 发现乳腺癌免疫细胞异质性和新靶点
乳腺癌精准治疗的一个主要障碍是人们对乳腺癌生态系统缺乏深入的了解。肿瘤生态系统由癌细胞、浸润免疫细胞、基质细胞和其他细胞类型以及非细胞组织成分组成[90]。由于遗传和非遗传因素,癌细胞和肿瘤相关细胞具有表型和功能的异质性。为了描述乳腺癌生态系统肿瘤细胞和免疫细胞的关系,Gerdes等[91]对乳腺导管癌组织免疫荧光切片进行单细胞分割,进行单细胞水平的定量以聚类方法确定了上皮细胞标志物和免疫标志物的共表达模式,发现上皮细胞聚集模式与免疫浸润的存在和类型有关,这表明每个患者的上皮肿瘤和免疫系统之间存在复杂的相互作用。这为分析上皮和免疫/基质成分对理解导管原位癌病变的复杂环境是必要的提供了第一个证据。毋庸置疑,乳腺肿瘤微环境中免疫细胞的鉴定在乳腺癌的治疗中尤为重要。随后,Wagner等[92]利用单细胞技术结合质谱分析对144例人乳腺肿瘤和50例非肿瘤组织样本测序分析,发现在肿瘤细胞组成上表现出明显的个体化差异,包括表型异常和表型显性。在ER+亚型中出现了较多的高表达PD-L1+的肿瘤相关巨噬细胞和衰竭T细胞,肿瘤细胞与免疫细胞的关系分析揭示了与免疫抑制和不良预后相关的生态系统特征。对肿瘤微环境中T细胞深入研究发现,其和正常组织中的T细胞相似,但肿瘤微环境特异性却在持续性的扩张。Azizi等[93]对8例乳腺癌患者27,000个T细胞和T细胞受体(T cell receptor, TCR)进行单细胞分析,揭示了TCR的利用对表型多样性的影响,证明了乳腺癌是T细胞持续活化的模型,与癌症中的巨噬细胞极化模型是不一致的。在肿瘤浸润性淋巴细胞中T细胞是占主导地位的,但是T细胞数量和差异与乳腺癌预后的关系尚不明确[94]。Savas等[95]也对6311个T细胞进行单细胞RNA测序,发现浸润的T 细胞群体存在明显的细胞异质性,其中CD8+T细胞的数量是最多的,并且具有组织驻留记忆T细胞(TRM)的分化特性,这些CD8+TRM细胞表达了高水平的免疫检查点分子和效应蛋白,与早期三阴性乳腺癌患者预后改善显著相关。
依据肿瘤微环境图谱,细胞大致可分化为肿瘤相关巨噬细胞、促血管生成细胞、原发肿瘤生长和远端转移细胞[96,97]。肿瘤相关巨噬细胞会通过抑制效应T细胞的分化和功能,刺激调节性T细胞和骨髓源性抑制细胞在肿瘤内积累,建立了一个强大的免疫抑制肿瘤生态系统[98~100]。最新研究发现,在乳腺癌小鼠模型中,钙/钙调蛋白依赖性蛋白激酶2 (calcium/calmodulin-dependent kinase kinase, CaMKK2)在瘤内髓样细胞内高表达,并证明其在髓样细胞内的抑制作用通过增加效应CD8+T细胞和免疫刺激髓样亚群的瘤内积累来抑制肿瘤生长。与对照组相比,从CaMKK2–小鼠中分离的肿瘤相关巨噬细胞表达了更高水平的参与效应T细胞招募的趋化因子。CaMKK2抑制剂可以抑制肿瘤以CD8+T细胞依赖的方式生长,并促进免疫细胞微环境的重新编程[101]。这些数据的发现使CaMKK2作为骨髓选择性检查点提供了依据,其抑制作用可能在乳腺癌的免疫治疗中发挥重要作用。
2.3.2 揭示乳腺癌耐药机制
化疗和靶向治疗耐药的出现是目前乳腺癌治疗面临的重大挑战。单细胞基因组和转录组测序强调了肿瘤内遗传异质性在肿瘤进化中的重要性,并表明未经治疗的肿瘤内遗传异质性是肿瘤耐药的关键因素[102~105]。然而,在许多情况下,驱动抗性的遗传机制尚未被发现,这提示非遗传机制在其中发挥了作用。单细胞转录和表观遗传机制研究在乳腺癌肿瘤细胞在面对环境、代谢或治疗压力的适应过程中发挥作用。Kim等[106]研究了TNBC患者肿瘤细胞对新辅助化疗(neoadjuvant chemotherapy, NAC)反应后的基因组和表型进化,发现了两种截然不同的克隆动力学:消退型和持续型。在克隆消退型患者中,NAC清除了肿瘤细胞,只留下正常的二倍体细胞类型,包括许多成纤维细胞和免疫细胞。与此相反,克隆持续型患者体内存在大量因NAC而改变基因型和表型的残余肿瘤细胞。之后利用单细胞DNA和RNA测序对8例TNBC患者进行深入分析,结果显示,TNBC患者在多西紫杉醇(docetaxel)和表阿霉素(epirubicin)治疗前后,其染色体数目和基因组发生了变化,并且被选择性的适应。治疗后患者的转录本进行了重编程,并且在治疗前已经表达了化学抗性基因的子集,这说明其耐药基因型是预先存在的,治疗后转录亚克隆的出现可能解释了癌细胞对治疗压力的适应。总的来说,该数据支持一个耐药模型,在这个模型中,两种模式的进化(适应性和获得性)正在建立耐药性肿瘤。
在基因转录表达的过程中,组蛋白修饰调控染色质结构是主要表观遗传机制和调控因子。相比单细胞转录组,表观遗传变异,如染色质特征,对肿瘤异质性和耐药进化的贡献仍然未知。最新研究,Grosselin等[77]则利用单细胞染色质免疫沉淀测序,在单细胞分辨率下描绘了成千上万个细胞的染色质景观,其依据细胞染色质分布情况对细胞类型进行分割,并识别了每个亚群的关键染色质特征。利用病人衍生的乳腺癌获得性耐化疗和靶向治疗的异种移植瘤模型,发现未经治疗的药物敏感肿瘤中有一部分细胞与耐药性肿瘤细胞共享一个共同的染色质特征。这些细胞,以及来自耐药性肿瘤的细胞,已经失去了染色质标记—H3K27me3,它与稳定的转录抑制相关,包括已知的促进化疗或靶向治疗的耐药基因。H3K27me3染色质标记的缺失可以使染色质转变为“允许”状态,并可能与转录变化之前的启动事件相对应。值得注意的是,在胰岛素样生长因子信号通路中存在H3K27去甲基化和胰岛素样生长因子结合蛋白(insulin-like growth factor binding protein, IGFBP)基因的转录激活,这在乳腺癌耐药性中起着关键作用。这项研究表明,具有耐药性肿瘤细胞染色质特征的罕见细胞在治疗前就已经存在。
随着乳腺癌肿瘤微环境图谱在不断的完善,乳腺癌肿瘤微环境中无数的肿瘤细胞和非恶性细胞,嵌入在富含糖蛋白的细胞外基质(extracellular matrix, ECM)中,主要的细胞类型包括内皮细胞、免疫细胞和癌相关成纤维细胞(cancer-related fibroblasts, CAFs)。ECM除了作为支持组织结构的物理支架,还是不同细胞类型之间的信号转换器,并且其病变的硬度和纤维胶原的丰度提供促进肿瘤发生和进展的机制信号,在乳腺癌中,基质细胞因子,尤其是那些与ECM重建相关的因子,对化疗反应有很强的预测作用[107~109]。肿瘤细胞会吸收微环境的基质成分以促进其进展,而肿瘤细胞命运则由激活转录级联响应发育信号通路的细胞外信号决定,如Hedgehog (Hh)信号通路、骨形态发生蛋白(bone morphogenetic protein, BMP)和成纤维细胞生长因子(fibroblast growth factor, FGF)[110,111]。Hh蛋白家族成员均由氨基端结构域(Hh-N)及羧基端结构域(Hh-C)组成,其中Hh-N有Hh蛋白的信号活性。乳腺癌主要表现为Hh-N依赖的通路激活,Hh-N与受体Ptc(Patched)结合,激活SMO (Smoothened),活化的SMO介导转录因子Gli1转位进入细胞核,驱动Hh靶基因的转录[112,113]。在乳腺癌不同亚型中Hh-N的表达被重新激活,尤其是预后差的三阴性乳腺癌(TNBC),30%的TNBC具有旁分泌Hh通路特征[114]。Cazet等[115]在TNBC动物模型中发现CAFs是对Hh-N刺激做出反应的初级基质细胞。激活的CAFs反过来为肿瘤细胞获得具有化学抗性的干细胞样表型提供了有利环境,为获得耐化疗的癌干细胞(cancer stem cell, CSC)表型提供支持。在小鼠模型和I期临床试验患者中,使用SMO小分子抑制剂(inhibitor of SMO, SMOi),如维莫德吉(Vismodegib)和索尼德吉(Sonidegib),提高了肿瘤细胞对多西紫杉醇(docetaxel)的化疗敏感性,降低了癌转移并提高了生存率(图2)。目前在TNBC动物模型中Hh信号以旁分泌的方式表达是很明显的,针对微环境耐受良好的药物种类有限,以及缺乏对微环境导向治疗反应的生物标志物,要将抗基质治疗纳入乳腺癌治疗的临床进展还须进行详细的研究。

图2 乳腺癌依赖Hh信号通路的耐药机制
静息状态下,受体Ptc与SMO不会相互作用(黑色停止箭头),当Hh-N与Ptc结合后,会激活SMO,活化的SMO介导转录因子Gli1入核,驱动靶基因转录,激活癌相关成纤维细胞,使肿瘤细胞获得药物抗性的干细胞样表型(黑色箭头)。SMOi抑制SMO活性,Gli1无法入核(红色停止箭头),解除肿瘤细胞药物抗性,提高对多西紫杉醇的化疗敏感性(红色箭头)。
3 结语与展望
单细胞技术的迅速发展及其在单个细胞遗传变异中的应用,极大地推动了生物医学的发展。尽管如此,目前该技术及其衍生技术仍存在其一定的缺陷,有着很大的改进潜力。在整个单细胞测序的技术流程中,获得单个具有活性的细胞样本是保证后续文库构建和测序数据质量的前提。目前单细胞捕获的方法各有利弊,可以依据研究目的来选择合适的实验技术路线和方法,但无论何种方法,保证单细胞的活性和完整性是单细胞技术成功的最关键因素。在单细胞文库构建的过程中,如何避免核酸丢失和扩增偏差,提高灵敏度和可重复性是其首要解决的问题[116]。可通过使用低吸附的离心管、添加DNase或 RNase抑制剂等以降低样本的损失[42]。但对于DNA二级结构和RNA的反转录效率,则需要寻找新的聚合酶以提高二级结构的扩增效率,或新的逆转录酶以提高逆转录效率[41]。由于单细胞测序技术的多样性,数据本身的稀疏和混乱,再加上材料样本的低质量,扩增过程及测序的偏差,使单细胞测序数据的分析变的更加困难,如何提高分析通量和节约成本,如何获得细胞的位置信息,尤其是测序数据中隐藏的非目标细胞,也是亟待解决的问题[116,117]。随着单细胞测序技术的广泛应用,细胞间的相关性和异质性逐渐变得清晰,反映在单细胞测序数据上的差异及如何与细胞功能相匹配是一个巨大的挑战,这不仅依赖于每个细胞中特定转录本的稳定性和半衰期,还与翻译后修饰的速率和特异性相关[118]。随着测序数据的海量增加,数据管理将成为一项复杂而繁琐的任务,并面临数据存储、挖掘分析、系统整合等诸多挑战。在未来,单细胞测序的多组学分析,能够对正常的细胞功能和疾病演变进行详尽的描述,因为其可以系统整合同一个单细胞的基因组、转录组、表观组或蛋白质组等分析,全面了解每个细胞,将克隆结构和细胞亚型与基因变异和表型直接联系起来。由于组织内的位置是细胞识别的关键决定因素,保存细胞的空间位置信息尤为重要,空间转录组的发展会逐渐揭开细胞时空特异性的面纱。单细胞(测序)技术可绘制全面的细胞类型参考地图,如果加上时间坐标,有利于在细胞层面绘制健康和疾病的全景图像,以及对疾病本质的深入理解,将为人们实现真正的精准医疗和个体化治疗奠定基础。
当前乳腺癌单细胞测序主要的问题集中在样本的收集和制备,因为多数的肿瘤样本都是经过冷冻,这样就不能保证细胞膜的完整性。同时,肿瘤组织在裂解成为单细胞悬液的过程中可能会发生细胞表面分子的改变。这些都会影响最终的数据分析,尤其是对每个细胞的功能注释。
基于单细胞测序技术以全面剖析乳腺癌的特征,已取得相当大的进步。单细胞亚克隆异质性的研究为乳腺癌亚克隆频率及其演变提供了新的细节,基本描述了乳腺肿瘤生态系统复杂的细胞表型,以及肿瘤生态系统各组成部分之间的关系,为人们提供了较为全面的乳腺癌生态系统的图谱。针对乳腺癌的单细胞测序显示拷贝数的进化发生在肿瘤进化的早期,而点突变随着时间的推移逐渐进化,产生更广泛的克隆多样性,为乳腺癌肿瘤进化的交叉分枝模型提供了支持。在乳腺癌个体患者中,非浸润性导管原位癌与邻近浸润性导管癌病变之间存在直接的谱系关系,大多数的突变和拷贝数变异(copy number variations, CNVs)侵袭之前已在导管内完成,并具有多个突变和CNVs克隆体从导管逃逸,共同迁移到邻近组织,形成侵袭性癌。乳腺癌的耐药性是由选择罕见的预先存在的克隆引起的,还是通过诱导新的突变产生获得性耐药性引起的一直备受争议。新辅助化疗前后乳腺癌患者的纵向样本的单细胞DNA测序表明,耐药基因型是预先存在的,是化疗选择的。尽管可以识别出表达与化疗耐药性相关的单个基因的细胞亚群,但具有耐药程序的细胞在治疗前无法检测到。这些数据表明,乳腺癌从发生到治疗,不断进化(演化)过程中存在明显的异质性,为人们更好地理解乳腺癌的发生、转移和耐药提供了证据。
对乳腺癌中同一个细胞整合基因组、表观基因组、转录组和/或蛋白质组的分析,可以全面了解每个细胞的功能和在疾病进化过程中的所扮演的角色。在未来,针对乳腺癌单细胞的多组学分析,并结合细胞分子成像等技术,从乳腺癌的遗传异质性、非遗传异质性,到耐药异质性,再到整个肿瘤微环境异质性,为绘制更加精确的乳腺癌细胞图谱提供支持,加深对乳腺癌演化规律的理解。同时,探索和发现乳腺癌发生、发展和转移中的分子标记,并为治疗干预提供候选的靶标分子,以及进一步开发精确的药物非常有益,为乳腺癌的免疫治疗提供新的策略。考虑到每个人的遗传(基因)、环境和生活方式等个体差异,单细胞技术(测序)有望实现乳腺癌的个体化诊治,对个体的早诊早治和预测预防具有重要的科学意义和应用价值。
[1] Yu TL, Cai DL, Zhu GF, Ye XJ, Min TS, Chen HY, Lu DR, Chen HM. Effects of CSN4 knockdown on proliferation and apoptosis of breast cancer MDA-MB-231 cells., 2019, 41(4): 318–326.余同露, 蔡栋梁, 朱根凤, 叶晓娟, 闵太善, 陈红岩, 卢大儒, 陈浩明. CSN4基因干扰对乳腺癌MDA-MB- 231细胞增殖和凋亡的影响. 遗传, 2019, 41(4): 318– 326.
[2] Boyle P, Levin B. World Cancer Report 2008. Lyon: IARC Press, 2008.
[3] Yao CB, Zhou X, Chen CS, Lei QY. The regulatory mechanisms and functional roles of the Hippo signaling pathway in breast cancer., 2017, 39(7): 617–629.姚传波, 周鑫, 陈策实, 雷群英. Hippo信号通路在乳腺癌中的调控机制及作用. 遗传, 2017, 39(7): 617– 629.
[4] Hanahan D, Weinberg RA. The hallmarks of cancer., 2000, 100(1): 57–70.
[5] Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation., 2011, 144(5): 646–674.
[6] Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, Butler A, Cheverton A, Gamble J, Hinton J, Jia M, Jayakumar A, Jones D, Latimer C, Lau KW, McLaren S, McBride DJ, Menzies A, Mudie L, Raine K, Rad R, Chapman MS, Teague J, Easton D, Langerød A, Lee MT, Shen CY, Tee BT, Huimin BW, Broeks A, Vargas AC, Turashvili G, Martens J, Fatima A, Miron P, Chin SF, Thomas G, Boyault S, Mariani O, Lakhani SR, van de Vijver M, van 't Veer L, Foekens J, Desmedt C, Sotiriou C, Tutt A, Caldas C, Reis-Filho JS, Aparicio SA, Salomon AV, Børresen-Dale AL, Richardson AL, Campbell PJ, Futreal PA, Stratton MR. The landscape of cancer genes and mutational processes in breast cancer., 2012, 486(7403): 400–404.
[7] Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple- negative breast cancers., 2012, 486(7403): 395– 399.
[8] Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A, Troge J, Ravi K, Esposito D, Lakshmi B, Wigler M, Navin N, Hicks J. Genome-wide copy number analysis of single cells., 2012, 7(6): 1024–1041.
[9] Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell., 2012, 338(6114): 1622– 1626.
[10] Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G1, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing., 2012, 366(10): 883–892.
[11] Yates LR, Campbell PJ. Evolution of the cancer genome., 2012, 13(11): 795–806.
[12] Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes., 2013, 339(6127): 1546–1558.
[13] Sethi N, Kang Y. Unravelling the complexity of metastasis—molecular understanding and targeted therapies.,2011, 11(10): 735–748.
[14] Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, Yue P, Zhang Y, Pant KP, Bhatt D, Ha C, Johnson S, Kennemer MI, Mohan S, Nazarenko I, Watanabe C, Sparks AB, Shames DS, Gentleman R, de Sauvage FJ, Stern H, Pandita A, Ballinger DG, Drmanac R, Modrusan Z, Seshagiri S, Zhang Z. The mutation spectrum revealed by paired genome sequences from a lung cancer patient., 2010, 465(7297): 473–477.
[15] Clark J, Attard G, Jhavar S, Flohr P, Reid A, De-Bono J, Eeles R, Scardino P, Cuzick J, Fisher G, Parker MD, Foster CS, Berney D, Kovacs G, Cooper CS. Complex patterns of ETS gene alteration arise during cancer development in the human prostate., 2008, 27(14): 1993–2003.
[16] Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, Michor F, Meric-Bernstam F, Navin NE. Clonal evolution in breast cancer revealed by single nucleus genome sequencing., 2014, 512(7513): 155–160.
[17] Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing., 2011, 472(7341): 90–94.
[18] Liu W, Hou YF, Chen HH, Wei HD, Lin WR, Li JC, Zhang M, He FC, Jiang Y. Sample preparation method for isolation of single-cell types from mouse liver for proteomic studies., 2011, 11(17): 3556– 3564.
[19] Dorrell C, Erker L, Lanxon-Cookson KM, Abraham SL, Victoroff T, Ro S, Canaday PS, Streeter PR, Grompe M. Surface markers for the murine oval cell response., 2008, 48(4): 1282–1291.
[20] Su XB, Shi Y, Zou X, Lu ZN, Xie GC, Yang JYH, Wu CC, Cui XF, He KY, Luo Q, Qu YL, Wang N, Wang L, Han ZG. Single-cell RNA-seq analysis reveals dynamic trajectories during mouse liver development., 2017, 18(1): 946.
[21] Der E, Ranabothu S, Suryawanshi H, Akat KM, Clancy R, Morozov P, Kustagi M, Czuppa M, Izmirly P, Belmont HM, Wang T, Jordan N, Bornkamp N, Nwaukoni J, Martinez J, Goilav B, Buyon JP, Tuschl T, Putterman C. Single cell RNA sequencing to dissect the molecular heterogeneity in lupus nephritis., 2017, 2(9): 93009.
[22] Autengruber A, Gereke M, Hansen G, Hennig C, Bruder D. Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function., 2012, 2(2): 112– 120.
[23] Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, Ryu JH, Wagner BK, Shen-Orr SS, Klein AM, Melton DA, Yanai I. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra- cell population structure., 2016, 3(4): 346– 360.e4.
[24] Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, van Oudenaarden A. A single-cell transcriptome atlas of the human pancreas., 2016, 3(4): 385–394.
[25] Wollny D, Zhao S, Everlien I, Lun XK, Brunken J, Brüne D, Ziebell F, Tabansky I, Weichert W, Marciniak- Czochra A, Martin-Villalba A. Single-cell analysis uncovers clonal acinar cell heterogeneity in the adult pancreas., 2016, 39(3): 289–301.
[26] Li D, Peng SY, Zhang ZW, Feng RC, Li L, Liang J, Tai S, Teng CB. Complete disassociation of adult pancreas into viable single cells through cold trypsin-EDTA digestion., 2013, 14(7): 596–603.
[27] Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq., 2014, 509(7500): 371–375.
[28] Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin α6β4 identifies an adult distal lung epithelial population with regenerative potential in mice., 2011, 121(7): 2855–2862.
[29] Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, Stripp BR, Whitsett JA. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis., 2016, 1(20): e90558.
[30] Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, Kasper M. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity., 2016, 3(3): 221–237.
[31] Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, McCarroll SA, Cepko CL, Regev A, Sanes JR. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics., 2016, 166(5): 1308–1323.
[32] Daniszewski M, Senabouth A, Nguyen QH, Crombie DE, Lukowski SW, Kulkarni T, Sluch VM, Jabbari JS, Chamling X, Zack DJ, Pébay A, Powell JE, Hewitt AW. Single cell RNA sequencing of stem cell-derived retinal ganglion cells., 2018, 5: 180013.
[33] Lafzi A, Moutinho C, Picelli S, Heyn H. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies., 2018, 13(12): 2742–2757.
[34] Gross A, Schoendube J, Zimmermann S, Steeb M, Zengerle R, Koltay P. Technologies for single-cell isolation.Int J Mol , 2015, 16(8): 16897–16919.
[35] Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell., 2012, 338(6114): 1622– 1626.
[36] Gole J, Gore A, Richards A, Chiu YJ, Fung HL, Bushman D, Chiang HI, Chun J, Lo YH, Zhang K. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells., 2013, 31(12): 1126.
[37] Grün D, Van Oudenaarden A. Design and analysis of single-cell sequencing experiments., 2015, 163(4): 799–810.
[38] Ellsworth DL, Blackburn HL, Shriver CD, Rabizadeh S, Soon-Shiong P, Ellsworth RE. Single-cell sequencing and tumorigenesis: improved understanding of tumor evolution and metastasis., 2017, 6(1): 15.
[39] Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers., 2014, 11(2): 163–166.
[40] Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells., 2017, 8: 14049.
[41] Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol , 2018, 19(3): 807.
[42] Liang JL, Cai WS, Sun ZH. Single-cell sequencing technologies: current and future., 2014, 41(10): 513–528.
[43] Hodne K, Weltzien FA. Single-cell isolation and gene analysis: pitfalls and possibilities.Int J Mol , 2015, 16(11): 26832–26849.
[44] Cheng L, Zhang S, MacLennan GT, Williamson SR, Davidson DD, Wang M, Jones TD, Lopez-Beltran A, Montironi R. Laser-assisted microdissection in translational research: theory, technical considerations, and future applications., 2013, 21(1): 31–47.
[45] Yao YX, La YF, Di R, Liu QY, Hu WP, Wang XY, Chu MX. Comparison of different single cell whole genome amplification methods and MALBAC applications in assisted reproduction., 2018, 40(8): 620–631.姚雅馨, 喇永富, 狄冉, 刘秋月, 胡文萍, 王翔宇, 储明星. 不同单细胞全基因组扩增方法的比较及MALBAC在辅助生殖中的应用. 遗传, 2018, 40(8): 620–631.
[46] Cai HQ, Liu HT, Shi B, Li A, Tang WR, Luo Y. Recent Advance of Whole Genome Amplification and its Application prospect in Forensic Individual Identification., 2010, 32(11): 1119–1125.蔡海强, 柳海涛, 史斌, 李安, 唐文如, 罗瑛. 全基因组扩增技术及其在法医个体识别中的应用. 遗传, 2010, 32(11): 1119–1125.
[47] Troutt AB, McHeyzer-Williams MG, Pulendran B, Nossal GJ. Ligation-anchored PCR: a simple amplification technique with single-sided specificity., 1992, 89(20): 9823–9825.
[48] Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: implications for genetic analysis., 1992, 89(13): 5847–5851.
[49] Telenius H, Carter NP, Bebb CE, Nordenskjöld M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide- primed PCR: general amplification of target DNA by a single degenerate primer., 1992, 13(3): 718–725.
[50] Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA., 1996, 93(25): 14676–14679.
[51] Dean FB, Hosono S, Fang LH, Wu XH, Faruqi AF, Bray-Ward P, Sun ZY, Zong QL, Du YF, Du J, Driscoll M, Song WM, Kingsmore SF, Egholm M, Lasken RS. Comprehensive human genome amplification using multiple displacement amplification., 2002, 99(8): 5261–5266.
[52] Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification., 2001, 11(6): 1095–1099.
[53] Lasken RS. Single-cell sequencing in its prime., 2013, 31(3): 211–212.
[54] Cohen AA, Geva-Zatorsky N, Eden E, Frenkel- Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen- Saidon C, Liron Y, Kam Z, Cohen L, Danon T, Perzov N, Alon U. Dynamic proteomics of individual cancer cells in response to a drug., 2008, 322(5907): 1511– 1516.
[55] Raj A, van Oudenaarden A. Single-molecule approaches to stochastic gene expression.,2009, 38: 255–270.
[56] Tang FC, Barbacioru C, Nordman E, Li B, Xu NL, Bashkirov VI, Lao KQ, Surani MA. RNA-Seq analysis to capture the transcriptome landscape of a single cell., 2010, 5(3): 516–535.
[57] Tang FC, Barbacioru C, Wang YZ, Nordman E, Lee C, Xu NL, Wang XH, Bodeau J, Tuch BB, Siddiqui A, Lao KQ, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell., 2009, 6(5): 377–382.
[58] Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity., 2013, 14(4): R31.
[59] Fan XY, Zhang XN, Wu XL, Guo HS, Hu YQ, Tang FC, Huang YY. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos., 2015, 16(1): 14.
[60] Sheng KW, Cao WJ, Niu YC, Deng Q, Zong CH. Effective detection of variation in single-cell transcriptomes using MATQ-seq., 2017, 14(3): 267–270.
[61] Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq., 2011, 21(7): 1160.
[62] Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S. Highly multiplexed and strand-specific single-cell RNA 5' end sequencing., 2012, 7(5): 813–828.
[63] Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2., 2014, 9(1): 171–181.
[64] Petersen M, Wengel J. LNA: a versatile tool for therapeutics and genomics., 2003, 21(2): 74–81.
[65] Vester B, Wengel J. LNA (Locked nucleic acid): high-affinity targeting of complementary RNA and DNA., 2004, 43(42): 13233–13241.
[66] Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets., 2015, 161(5): 1202–1214.
[67] Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq., 2011, 21(7): 1160–1167.
[68] Hochgerner H, Lönnerberg P, Hodge R, Mikes J, Heskol A, Hubschle H, Lin P, Picelli S, La Manno G, Ratz M, Dunne J, Husain S, Lein E, Srinivasan M, Zeisel A, Linnarsson S. STRT-seq-2i: dual-index 5′ single cell and nucleus RNA-seq on an addressable microwell array., 2017, 7(1): 16327.
[69] Soumillon M, Cacchiarelli D, Semrau S, van Oudenaarden A, Mikkelsen TS. Characterization of directed differentiation by high-throughput single-cell RNA-Seq., 2014, https://doi.org/10.1101/003236.
[70] Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification., 2012, 2(3): 666–673.
[71] Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole- organism science., 2013, 14(9): 618–630.
[72] Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li SQ, Livak KJ, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I. CEL- Seq2: sensitive highly-multiplexed single-cell RNA-Seq., 2016, 17: 77.
[73] Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell Transcriptomics applied to embryonic stem cells., 2015, 161(5): 1187–1201.
[74] Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types., 2014, 343(6172): 776–779.
[75] Guo HS, Zhu P, Wu XL, Li XL, Wen L, Tang FC. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing., 2013, 23(12): 2126–2135.
[76] Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W, Kelsey G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity., 2014, 11(8): 817–820.
[77] Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F, Frenoy O, Pousse Y, Reichen M, Woolfe A, Brenan C, Griffiths AD, Vallot C, Gérard A. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer., 2019, 51(6): 1060– 1066.
[78] Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation., 2015, 523(7561): 486–490.
[79] Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing., 2015, 348(6237): 910–914.
[80] Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, Mollbrink A, Linnarsson S, Codeluppi S, Borg A, Pontén F, Costea PI, Sahlén P, Mulder J, Bergmann O, Lundeberg J, Frisén J. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics., 2016, 353(6294): 78–82.
[81] Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi XQ, Chen K, Scheet P, Vattathil S, Liang H, Multani A, Zhang H, Zhao R, Michor F, Meric-Bernstam F, Navin NE. Clonal evolution in breast cancer revealed by single nucleus genome sequencing., 2014, 512(7513): 155–160.
[82] Gao RL, Kim C, Sei E, Foukakis T, Crosetto N, Chan LK, Srinivasan M, Zhang H, Meric-Bernstam F, Navin N. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer., 2017, 8(1): 228.
[83] Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, Kan Z, Han W, Park WY. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer., 2017, 8: 15081.
[84] Allred DC, Wu Y, Mao S, Nagtegaal ID, Lee S, Perou CM, Mohsin SK, O’Connell P, Tsimelzon A, Medina D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution., 2008, 14(2): 370–378.
[85] Virnig BA, Tuttle TM, Shamliyan T, Kane RL. Ductal carcinoma in situ of the breast: a systematic review of incidence, treatment, and outcomes.J Nati Inst, 2010, 102(3): 170–178.
[86] Gao RL, Davis A, McDonald TO, Sei E, Shi XQ, Wang Y, Tsai PC, Casasent A, Waters J, Zhang H, Meric- Bernstam F, Michor F, Navin NE. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer., 2016, 48(10): 1119–1130.
[87] Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple- negative breast cancers., 2012, 486(7403): 395– 399.
[88] Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li Y, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, Maetens M, Ignatiadis M, Stratton MR, Sotiriou C, Richardson AL, Lønning PE, Wedge DC, Campbell PJ. Subclonal diversification of primary breast cancer revealed by multiregion sequencing., 2015, 21(7): 751–759.
[89] Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, Casasent T, Meric-Bernstam F, Edgerton ME, Navin NE. Multiclonal invasion in breast tumors identified by topographic single cell sequencing., 2018, 172(1–2): 205–217.e12.
[90] McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship., 2010, 28(26): 4022–4028.
[91] Gerdes MJ, Gökmen-Polar Y, Sui Y, Pang AS, LaPlante N, Harris AL, Tan PH, Ginty F, Badve SS. Single-cell heterogeneity in ductal carcinoma in situ of breast., 2018, 31(3): 406–417.
[92] Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst S, Soysal SD, Jacobs A, Windhager J, Silina K, van den Broek M, Dedes KJ, Rodríguez Martínez M, Weber WP, Bodenmiller B. A single-cell atlas of the tumor and immune ecosystem of human breast cancer., 2019, 177(5): 1–16.
[93] Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, Choi K, Fromme RM, Dao P, McKenney PT, Wasti RC, Kadaveru K, Mazutis L, Rudensky AY, Pe'er D. Single-Cell Map of diverse immune phenotypes in the breast tumor microenvironment., 2018, 174(5): 1293–1308.e36.
[94] Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES, Coussens LM. Leukocyte composition of human breast cancer., 2012, 109(8): 2796– 2801.
[95] Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Kathleen, Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis., 2018, 24(7): 986–993.
[96] Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy., 2014, 41(1): 49–61.
[97] Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis., 2010, 141(1): 39–51.
[98] Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells., 2014, 26(5): 623–637.
[99] Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor- induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages., 2015, 125(9): 3365–3376.
[100] Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation., 2009, 86(5): 1065–1073.
[101] Racioppi L, Nelson ER, Huang W, Mukherjee D, Lawrence SA, Lento W, Masci AM, Jiao Y, Park S, York B, Liu YP, Baek AE, Drewry DH, Zuercher WJ, Bertani FR, Businaro L, Geradts J, Hall A, Means AR, Chao N, Chang CY, McDonnell DP. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer., 2019, 10(1): 2450.
[102] Roth A, McPherson A, Laks E, Biele J, Yap D, Wan A, Smith MA, Nielsen CB, McAlpine JN, Aparicio S, Bouchard-Côté A, Shah SP. Clonal genotype and population structure inference from single-cell tumor sequencing., 2016, 13(7): 573–576.
[103] Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, Shlien A, Cooke SL, Hinton J, Menzies A, Stebbings LA, Leroy C, Jia M, Rance R, Mudie LJ, Gamble SJ, Stephens PJ, McLaren S, Tarpey PS, Papaemmanuil E, Davies HR, Varela I, McBride DJ, Bignell GR, Leung K, Butler AP, Teague JW, Martin S, Jönsson G, Mariani O, Boyault S, Miron P, Fatima A, Langerød A, Aparicio SA, Tutt A, Sieuwerts AM, Borg Å, Thomas G, Salomon AV, Richardson AL, Børresen- Dale AL, Futreal PA, Stratton MR, Campbell PJ. The life history of 21 breast cancers., 2012, 149(5): 994–1007.
[104] McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future., 2017, 168(4): 613–628.
[105] Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies., 2018, 15(2): 81–94.
[106] Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, Crosetto N, Foukakis T, Navin NE. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing., 2018, 173(4): 879–893.e13.
[107] Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation., 2011, 144(5): 646–674.
[108] Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling., 2009, 139(5): 891–906.
[109] Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression., 2016, 22(5): 497–505.
[110] Perrimon N, Pitsouli C, Shilo BZ. Signaling mechanisms controlling cell fate and embryonic patterning., 2012, 4(8): a005975.
[111] Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer., 2002, 296(5570): 1046–1049.
[112] Hui M, Cazet A, Nair R, Watkins DN, O'Toole SA, Swarbrick A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy., 2013, 15(2): 203.
[113] Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer., 2013, 19(11): 1410–1422.
[114] O'Toole SA, Machalek DA, Shearer RF, Millar EK, Nair R, Schofield P, McLeod D, Cooper CL, McNeil CM, McFarland A, Nguyen A, Ormandy CJ, Qiu MR, Rabinovich B, Martelotto LG, Vu D, Hannigan GE, Musgrove EA, Christ D, Sutherland RL, Watkins DN, Swarbrick A. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer., 2011, 71(11): 4002–4014.
[115] Cazet AS, Hui MN, Elsworth BL, Wu SZ, Roden D, Chan CL, Skhinas JN, Collot R, Yang J, Harvey K, Johan MZ, Cooper C, Nair R, Herrmann D, McFarland A, Deng N, Ruiz-Borrego M, Rojo F, Trigo JM, Bezares S, Caballero R, Lim E, Timpson P, O'Toole S, Watkins DN, Cox TR, Samuel MS, Martín M, Swarbrick A. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer., 2018, 9(1): 2897.
[116] Wen L, Tang FC. Recent progress in single-cell RNA-Seq analysis., 2014, 36(11): 1069–1076.文路, 汤富酬. 单细胞转录组高通量测序分析新进展. 遗传, 2014, 36(11): 1069–1076.
[117] Kulkarni A, Anderson AG, Merullo DP, Konopka G. Beyond bulk: a review of single cell transcriptomics methodologies and applications., 2019, 58: 129–136.
[118] Hedlund E, Deng QL. Single-cell RNA sequencing: Technical advancements and biological applications., 2018, 59: 36–46.
Single-cell sequencing and its application in breast cancer
Qiang Zhang, Mingliang Gu
Breast cancer originates from ducts and epithelial cells, and gradually develops from hyperplasia to atypical hyperplasia,(adeno) carcinoma, to early and advanced invasive carcinoma. Traditional high-throughput sequencing mainly aims to identify candidate ‘driver genes’ attributable to development and progression of breast cancer, which has deficiencies in characterizing genomic structure alteration and subclone evolution, and thus ignores intratumoral, intertumoral or interpatient heterogeneity. The single-cell sequencing technology analyzes transcriptome (e.g., gene copy number and gene expression), explores cellular composition, differentiation and fate, fine-maps the tumor microenvironment, and provides supporting evidence for accurate stratification as well as personalized, precise therapy. At the same time, a complex relationship between breast cancer cells and T cells, macrophages and other immune cells can be revealed, thus facilitating discovery of new therapeutic targets and immune checkpoints. Here, we review state-of-the-art single-cell sequencing technologies and its application in breast cancer, in order to decipher multi-faceted alterations in the crosstalk/interactions between tumors and its microenvironments at the single-cell level, and provide a basis for better understanding of complicated pathogenesis and new avenues for immunotherapy.
single-cell sequencing; breast cancer; tumor microenvironment; immunotherapy
2019-10-25;
2020-02-03
国家重点研发计划“干细胞及转化研究”重点专项(编号:2018YFA0109500)和山东省自然科学基金青年基金项目(编号:ZR2019QH009)资助[Supported by the National Key R&D Program of China (No.2018YFA0109500), and the Youth Fund of Natural Science Foundation of Shandong Province (No. ZR2019QH009)]
张强,硕士,实习研究员,研究方向:分子遗传学。E-mail: qiangzhang17@yeah.net
顾明亮,硕士,特聘教授,研究方向:遗传学与基因组学。E-mail: minglianggu@hotmai.com; guml@big.ac.cn
10.16288/j.yczz.19-268
2020/1/17 17:18:05
URI: http://kns.cnki.net/kcms/detail/11.1913.r.20200117.0949.002.html
(责任编委: 孙玉洁)
猜你喜欢
临床肝胆病杂志(2022年8期)2022-11-23
中华骨与关节外科杂志(2022年1期)2022-08-31
中国临床医学影像杂志(2022年6期)2022-07-26
今日农业(2021年11期)2021-08-13
现代企业(2021年2期)2021-07-20
中国生殖健康(2020年4期)2020-12-09
科学(2020年4期)2020-11-26
中西医结合肝病杂志(2020年2期)2020-10-27
教育界·上旬(2020年8期)2020-06-27
中成药(2018年7期)2018-08-04
