
74-plex SNPs复合检测体系在中国人群中的族群推断研究
2020-04-15 03:20刘杨孙昌春马咪王玲赵雯婷马泉季安全刘京李彩霞
遗传 2020年3期
刘杨,孙昌春,马咪,王玲,赵雯婷,马泉,季安全,刘京,李彩霞
研究报告
74-plex SNPs复合检测体系在中国人群中的族群推断研究
刘杨1,2,孙昌春1,2,马咪2,3,王玲2,赵雯婷2,马泉2,季安全2,刘京2,李彩霞1,2
1. 山西医科大学,太原 036000 2. 公安部物证鉴定中心,法医遗传学公安部重点实验室,现场物证溯源技术国家工程实验室,北京 100038 3. 新疆生产建设兵团第七师公安局,奎屯 833200
使用一组祖源SNP可以分析某人群的遗传成分,推断某个体的族群来源。本课题组前期筛选出74个SNP位点实现了撒哈拉以南的非洲、北非、欧洲、美洲、大洋洲、南亚、西南亚、东亚、东北亚和东南亚等10个地理区域人群的推断,并基于MassARRAY质谱分析技术构建了74-plex SNPs复合检测体系。本研究利用该体系对14个中国人群1371份样本进行基因分型,验证评估该体系对中国人群的区分能力和法医学应用效能。首先,基于全球57个人群3628份个体构建参考人群分型库,采用Structure分析和等位基因频率热图等方法进行人群区分能力评估;然后,选取千人基因组计划中3个人群(不包含在参考人群分型库中)及本实验室检测的14个人群共计1654个体作为测试数据集,通过似然比和族群成分等统计分析,评估该体系对实际样本的族群来源推断能力。结果表明,DNA的量最低为1.5 ng时,74个SNP均可正确判型,适用于微量检材的检测;该体系对全球10个地理区域人群有区分能力,针对测试人群中欧洲、美洲、南部非洲个体族群来源推断的准确率为95.4%、不排除率为1.06%,东亚个体推断的准确率为71.0%、不排除率为17.9%,东南亚个体推断的准确率66.4%、不排除率为 33.3%。该方法可以为实际案件侦察提供线索。
法医遗传学;祖源SNP;族群推断;中国人群
DNA供者的族群地域分析不仅对于生物医药、人类迁移进化等研究有重要参考价值,而且在法 庭科学领域也具有重要应用价值,近年来被广泛关注[1~4]。当犯罪嫌疑人遗留在现场生物检材的STR (short tandem repeat, STR)数据与DNA数据库或者某个嫌疑人没有比中时,如果能够对生物检材来源人的族群、地域进行推断,将有助于锁定嫌疑人范围,促进案件定性和明确侦查方向。通过测序技术获得个人基因组上的祖源SNP (ancestry informative SNPs, AISNPs)分型信息,比较这些SNP分型数据与参考族群的相似性,可以计算族源成分,推断其族群来源[5~9]。
目前报道了大量洲际人群区分的AISNPs体系[10~14]。本课题组前期筛选出的74个SNP位点能够实现全球10个地理区域人群(撒哈拉以南的非洲、北非、欧洲、美洲、大洋洲,南亚、西南亚、东亚、东北亚、东南亚)的区分[15],且基于质谱技术构建了74-plex SNPs复合检测体系[16],实现了东亚人群的南北方遗传成分的进一步区分。但是,尚未进行该复合检测体系的性能验证及大规模样本的验证。本文利用74-plex SNPs复合检测体系对14个中国人群1371份样本进行基因分型,并对74-plex SNPs复合检测体系进行了体系性能验证和大规模样本的区分能力验证。本研究的成果可进一步丰富我国人群的AISNPs位点的数据,进而为中国不同语系人群特异性位点的筛选打下基础,并且可以为案件提供侦查线索。
1 材料与方法
1.1 样本信息
参考数据库参照前期文献报道[17],共计57个人群3628份个体。另外选取千人基因组3个人群(不包含在参考数据库中)和本实验室检测的14个中国人群,共17个人群1654份个体作为测试样本。基于参考数据库进行族群来源推断,评估体系在实际样本中的族群来源区分能力。本实验室样本均来源于国家科技资源共享服务平台计划项目(编号:YCZYPT[2017]01-3)。测试人群样本详细信息见表1。本实验室检测的所有样本对象均签署知情同意书及自述其详细族群信息。本研究已通过公安部物证鉴定中心伦理委员会的审查批准。
1.2 DNA的提取和定量
静脉血样本DNA的提取采用德国QIAGEN公司QIAamp®DNA Blood Midi试剂盒;用NanoDrop 2000C分光光度计(Thermo Scientific公司,美国)进行定量。用18.2MΩ去离子灭菌水调整浓度至5~ 10 ng/μL备检。
1.3 SNP位点来源
74个SNP位点源于本课题组前期筛选[15],基于MassARRAY质谱检测平台构建了复合检测体系[16],74个SNP位点在3个反应孔中检测,SNP位点信息见表2。
1.4 检测SNP分型
PCR复合扩增及纯化:PCR复合扩增反应体系为5 μL,PCR反应条件:95℃ 2 min,95℃ 30 s,56℃ 30 s,72℃ 1 min,循环45次,最后延伸72℃ 5 min。纯化反应体系为7 μL,充分振荡混匀后37℃孵育40 min,85℃ 5 min灭活酶活性。
单碱基延伸反应:采用9 μL体系, 94℃ 30 s;94℃ 5 s,(52℃ 5 s和80℃ 5 s,循环5次),共40个循环;然后72℃ 3 min。
树脂纯化:延伸后的体系加15 mg的Clean Resin树脂进行脱盐纯化。将Clean Resin树脂平铺到树脂板中,将干燥后的树脂倒入延伸产物板中,封膜,低速垂直旋转25 min使树脂与反应物充分接触,3000 r/min离心5 min使树脂沉入孔底部。
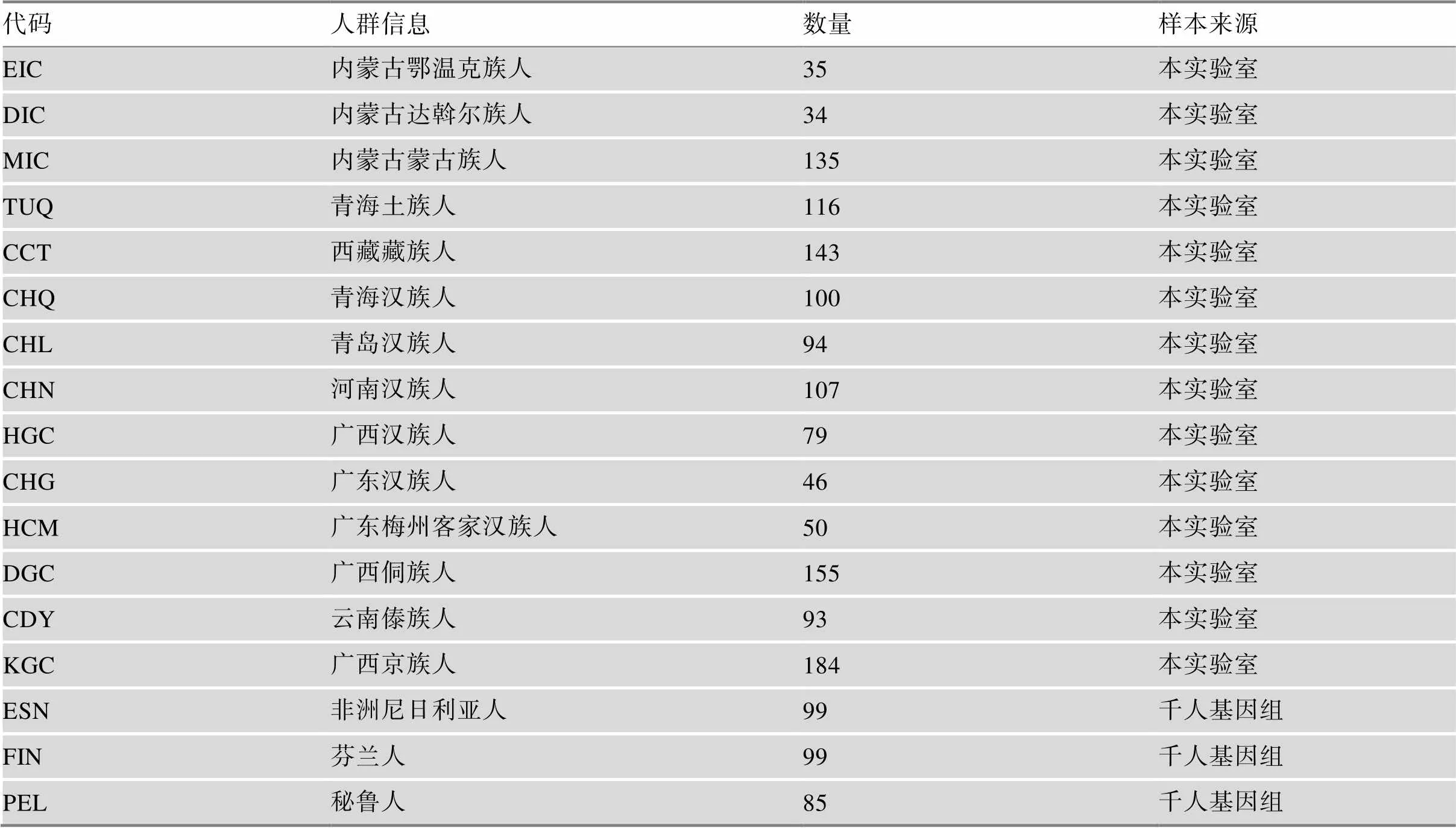
表1 测试人群样本信息表
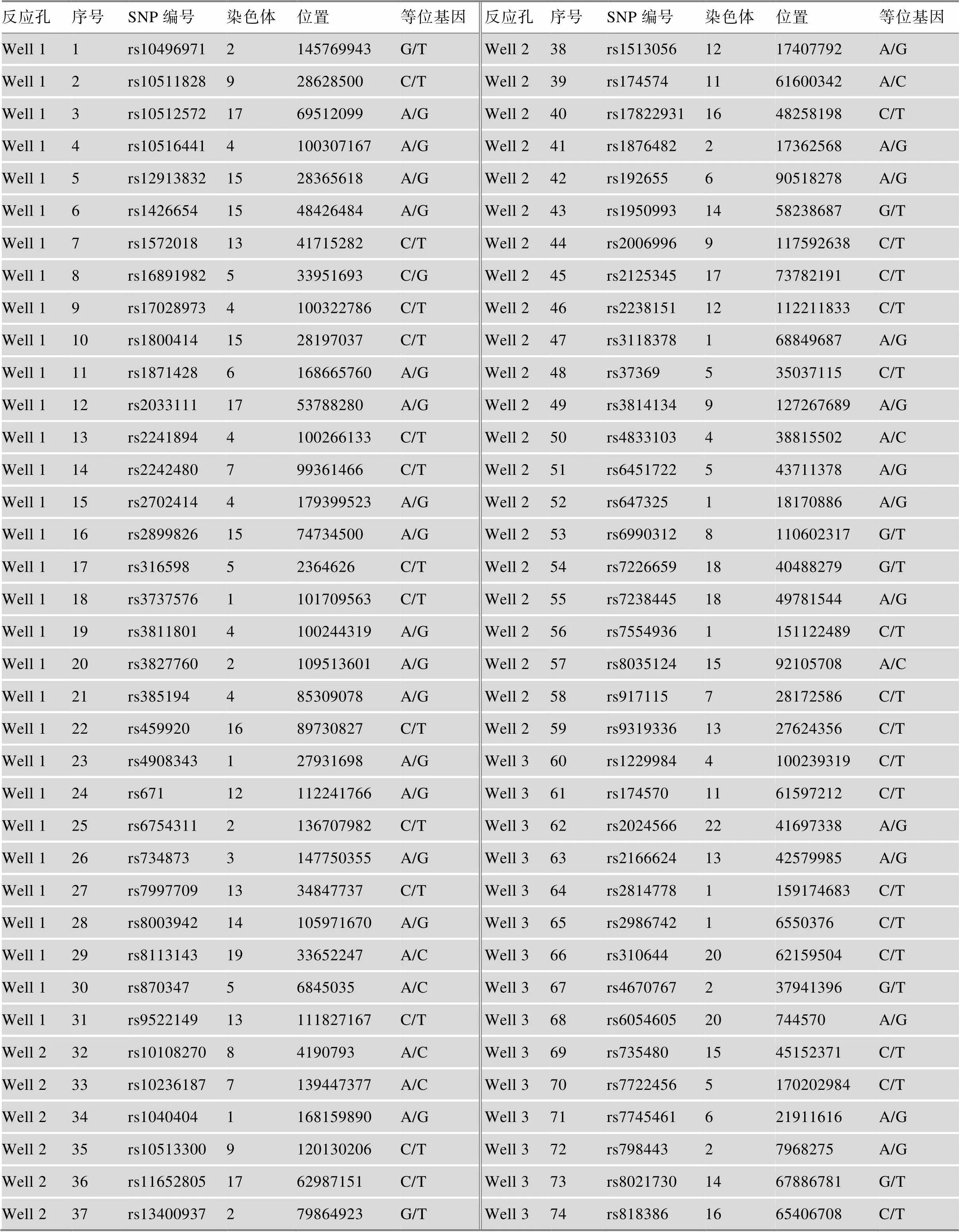
表2 每个反应孔中的SNP位点信息
芯片点样和质谱检测:用点样仪(MassARRAYTM Nanodispenser RS1000,美国Agena公司)把纯化后的样本点到带有基质的芯片上(8~15 nL)。然后用质谱检测分析仪(MassARRAYTM Analyzer,美国Agena公司)进行分型检测[18]。用TYPER 4.0软件对分型结果进行分析。
1.5 性能指标验证
分型准确性验证:选取5份样本:9947A、B0242、LCX、QEF、U144送至生工生物工程有限公司进行Sanger测序,验证本研究检测体系的基因分型与测序结果一致性。
灵敏度验证:将10 ng/μL标准品9947做浓度梯度稀释,15 μL体系中DNA 模板最终量分别为30、15、6、3、1.5和0.6 ng。使用构建的74-plex SNPs复合检测体系进行扩增和基因分型,每个浓度重复3次,用于验证该检测体系的灵敏度。
1.6 分析方法
1.6.1 Structure分析
针对全球10大区域人群分型数据库,用Structure 2.3.4[19]软件进行族群成分分析(=3-10,run=15, 10000 burnins,10000 MCMC), 分析各人群的遗传结构。使用Clumpak软件绘制Structure结果人群聚类图,相似度的阈值设置为0.9。
1.6.2 等位基因频率热图分析
用Genepop软件(http://www.genepop.curtin.edu. au/genepop_op5.html)计算每个位点的等位基因频率,使用R v3.0.1软件绘制等位基因频率热图。
1.6.3 群体匹配概率和似然比
用DNA族群推断系统软件(DAA)[20]计算17个人群1654份测试样本的群体匹配概率(AMP)和似然比(LR),当LR>10时,AMP排第一位的人群为未知个体的来源族群,当LR≤10时,AMP排序前两位人群均不排除。
1.6.4 箱形图分析
用Structure软件分析17个人群1654份测试样本的族群成分(=10, run=15),基于每个个体族群成分的最大值、最小值、中位数和两个四分位数,用EXCLE2016软件绘制箱线图展示每个个体族群成分的分布。
2 结果与分析
2.1 74-plex SNPs复合检测体系性能指标验证结果
分型准确性验证:5份测序样本共获得370个SNP分型数据,经对比测序结果与本研究复合检测体系所获得的基因分型100%一致。
灵敏度验证:使用构建的74-plex SNPs复合检测体系检测模板量为30~0.6 ng的9947。3次重复结果均显示,DNA模板量最低为1.5 ng时74个位点等位基因均可正确判型(图1)。
2.2 用全球10大区域人群分型数据库对体系效能进行评价
2.2.1 Structure族群成分分析结果
图2展示了全球57个人群3628个个体的Structure分析结果(=3~10),图中展示的是每个值多次运算结果中的最主要的聚类模式。当=10时,57个人群被聚类为撒哈拉以南的非洲、北非、西南亚、欧洲、南亚、东亚、东北亚、东南亚、大洋洲和美洲等10个区域。
2.2.2 等位基因频率热图
基于57个人群在74个SNP位点的等位基因频率分布,绘制等位基因频率聚类热图(图3)。通过图3可以找出人群特异SNP位点,例如rs10108270、rs2986742、rs7238445和rs451722聚类在一起,且它们在南非人群中的频率明显高于其他人群,说明这些位点分型是南非人群特异位点。57个人群在热图的左侧聚为10簇,分别为撒哈拉以南的非洲、北非、欧洲、美洲、大洋洲、南亚、西南亚、东亚、东北亚和东南亚。
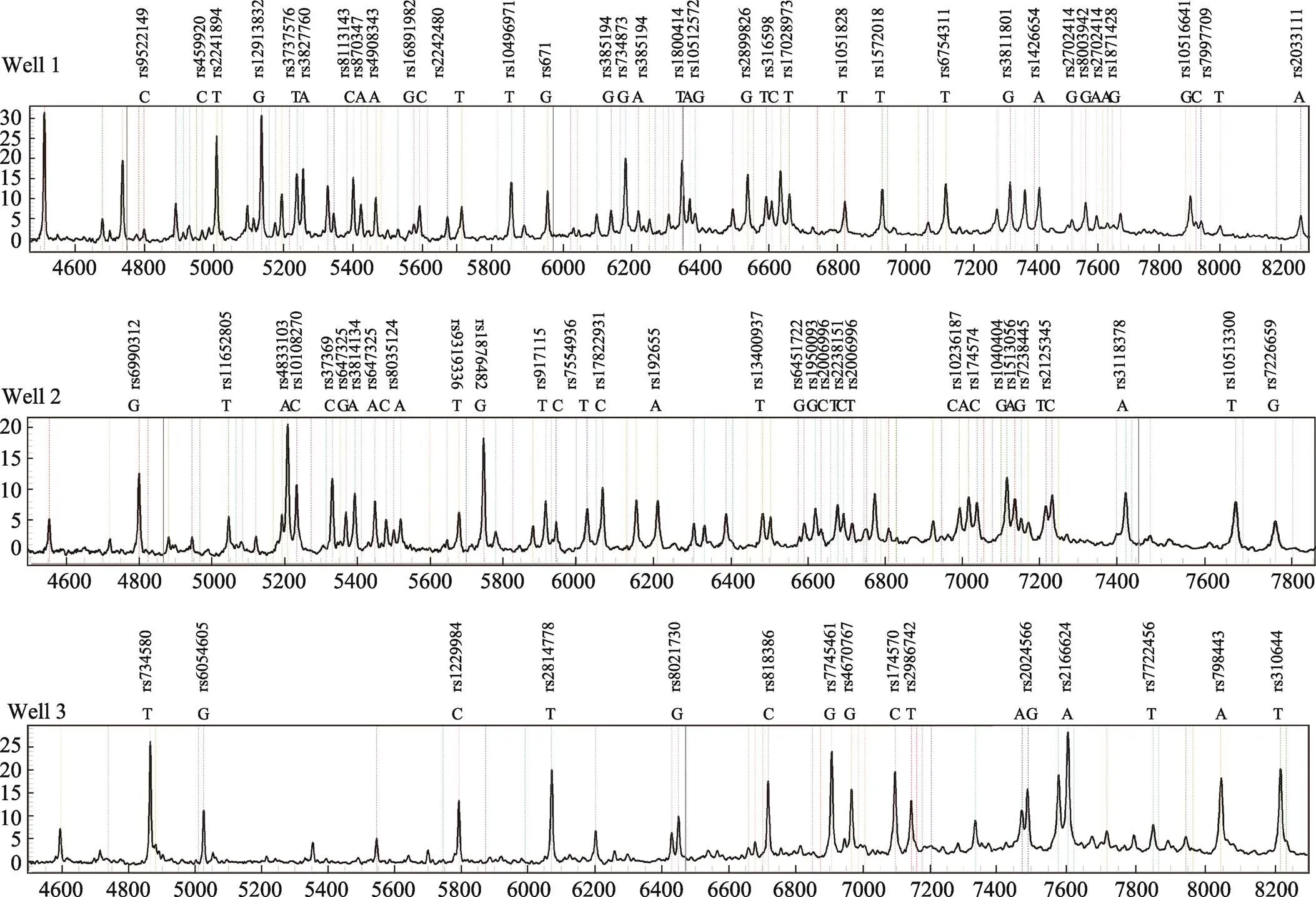
图1 DNA模板量为1.5 ng的分型结果
2.3 17个人群1654份测试个体的族群来源推断
本文使用17个人群1654份个体作为测试数据集评估74-plex SNPs复合检测体系的族群来源推断能力,验证体系在实际样本中的应用效能。所有测试样本均不包括在参考数据库中。
2.3.1 似然比
对已知来源的1654份样本基于参考数据库进行随机人群匹配概率计算,基于似然比进行族群来源的统计如表3。对测试人群整体的推断准确率为74%,不排除率为19%,错误率为7%。针对测试人群中欧洲、美洲、南部非洲个体族群来源推断的准确率为95.4%,不排除率为1.06%;东亚个体推断的准确率为71.0%,不排除率为17.9%,错误率为11.1%;东南亚个体推断的准确率66.4%,不排除率为33.3%,错误率为0.2%。
2.3.2 族群成分
对已知来源的1654份样本基于参考数据库使用Structure 2.3.4软件计算其族群成分(=10, run=10)。统计每个人群的平均族群成分见表3,所有样本的族群成分绘制箱线图(图4,A和B)。表3可见内蒙古蒙古族(MIC)、达斡尔族(DIC)、和鄂温克族(EIC)人群的东北亚成分的平均值分别为0.56、0.45和0.31;东亚成分为0.31、0.42和0.56。西藏藏族(CTT)和青海土族(TUQ)人群以东北亚成分为主,分别为0.78和0.63。青海汉族(CHQ)表现为东北亚和东亚成分的混合,族群成分平均值分别0.46和0.41。青岛汉族(CHL)和河南汉族(CHN)人群的以东亚成分为主,族群成分平均值分别为0.61和0.65。广西汉族(HGC)、广东客家汉族(HCM)和广东汉族(CHG)等中国南方汉族人群中东亚成分的平均值分别为0.31、0.38和0.49;东南亚成分的平均值分别为0.6、0.53和0.45。广西京族(KGC)、广西侗族(DGC)和云南傣族(CDY)等中国南方少数民族以东南亚成分为主,3个人群东南亚成分的平均值均大于0.6。
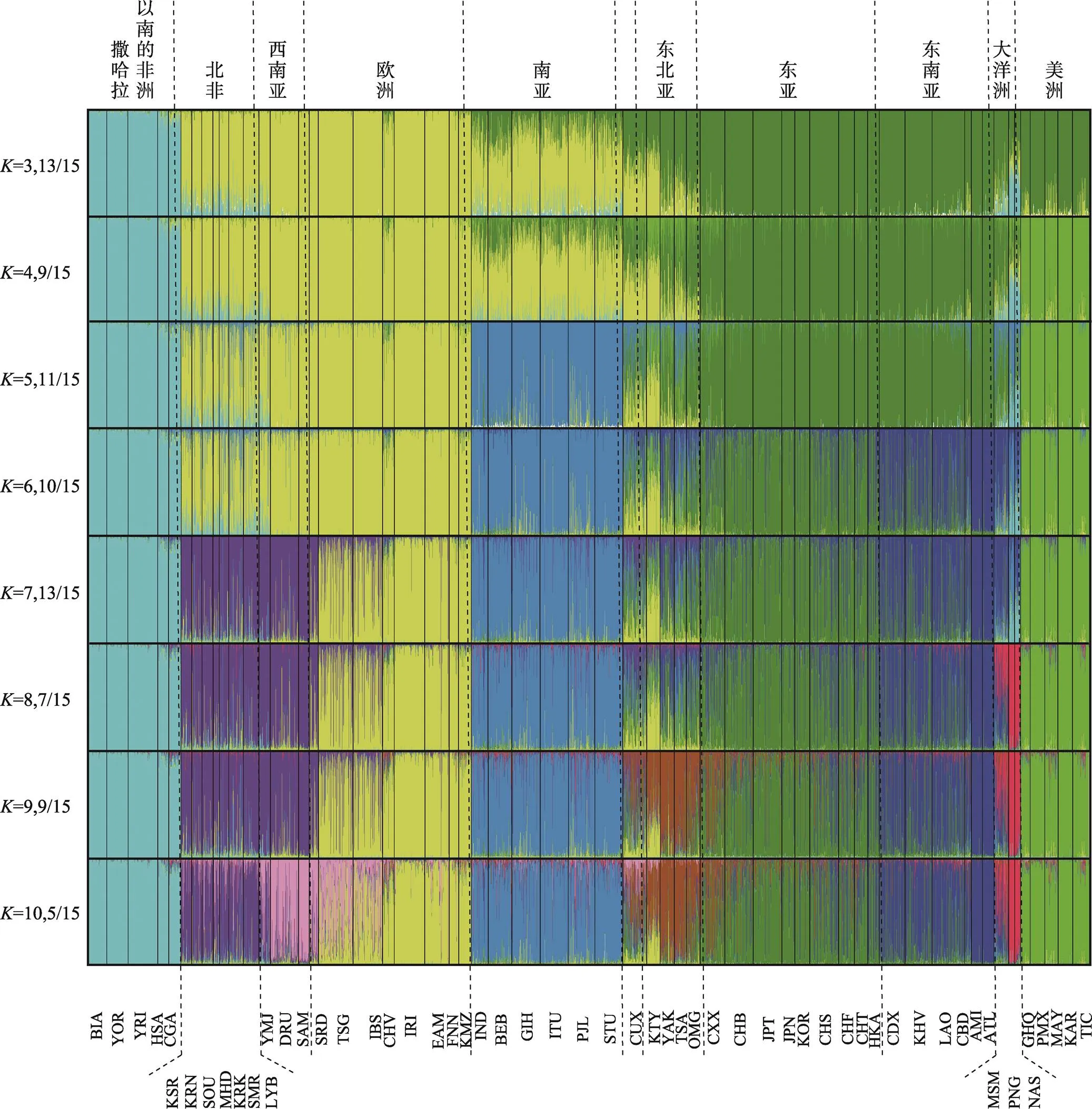
图2 74 SNPs的57人群Structure分析结果
值是指运行Structure分析时,用户假定全部群体分为几个亚群, 每个亚群在图中用一个单独颜色表示, 图中展示的是每个值多次运算结果中的最主要的聚类模式, 例如:当=3时,15次运算中有13次结果中的祖先成分模式为图2中所示模式。
3 讨论
本课题组前期建立了针对3大洲际人群(亚、欧、非)的27-plex SNPs和五大洲际人群的28-plex SNPs,实现了洲际人群的区分及其遗传结构的分析,但对洲际人群内部亚人群的进一步区分效力不足。而近年来,相关研究已经逐步从洲际群体鉴别过渡到亚人群的鉴别研究,如Phillips的“MAPlex”体系[21]使用164个遗传标记实现了非洲、中东、欧洲、南亚、东亚、美洲、大洋洲人群的区分,但该组位点没有实现东亚人群的进一步细分;Sun等[22]的12个multi-indels推断体系实现了喀拉拉人(Keralites)、老挝人(Laotians)、日本人(Japanese)、汉族(Han)和中国藏族(Chinese Tibetan)人群的区分。本研究体系实现了东南亚、东亚与东北亚人群的进一步区分,以及北非、西南亚与欧洲人群的进一步区分,且构建了适用于法医现场生物物证的检测体系,在实际应用中,可以使案件现场遗留的生物物证的族群来源进一步细化。在下一步研究中可以借鉴12个Multi- indels推断体系[22]等相关研究中的位点,构建更加精细的针对东亚人群的区分体系。

图3 74个SNPs的57个人群等位基因频率热图
颜色的深浅代表SNP位点的基因频率在不同群体中的相似性和差异性,红色表示最高等位基因频率,蓝色表示最低等位基因频率。
3.1 体系性能验证
5份样本的Sanger测序结果与本研究的74-plex SNPs复合检测体系检测的SNP分型100%一致。灵敏度结果显示,模板量最低为1.5 ng时74个位点等位基因均可正确判型,适用于微量检材的检测。该体系尚未进行检材适应性、组织统一性的验证,后期需要进行该两项的测试。
3.2 人群的遗传区分能力
本研究是基于全球十个区域57个人群为参考数据库进行族群来源分析,与本课题组2016年研究的61个参考人群相比做了以下优化:(1)增加了维吾尔族(CUX)和锡伯族(CXX),以评估该体系在新疆人群中的区分能力;(2)为避免人群样本数量不均一带来的结果偏差,将样本量较少且遗传结构相近的群体进行了合并,(比如,欧洲人群中的TSI和GRK人群合并为TSG,南亚人群中的KER、THT和KCH人群合并为IND,大洋洲人群中的MLY、SMO和MCR人群合并为MSM,美洲人群中的GHB和QUE人群合并为GHQ)。
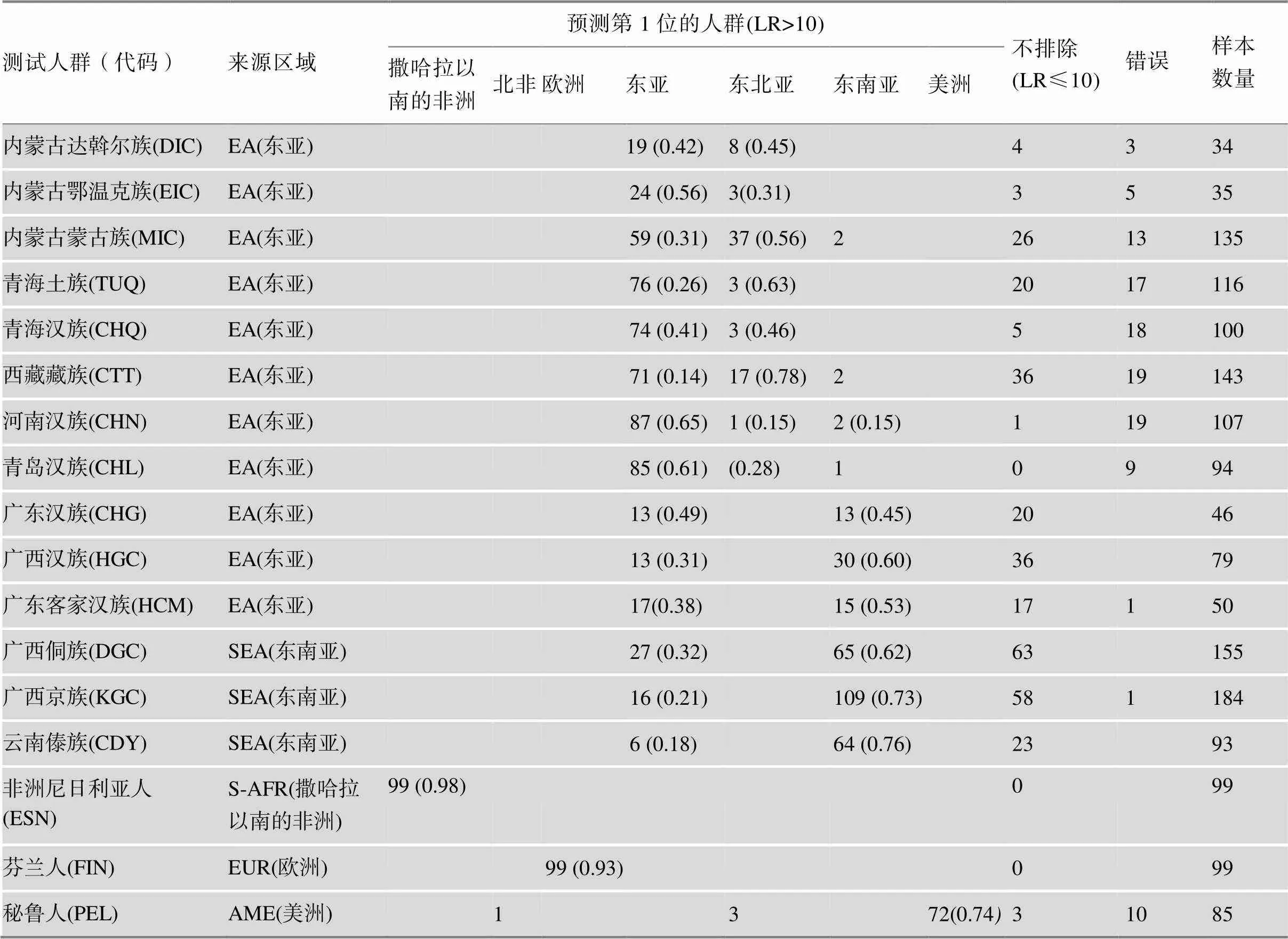
表3 测试样本的族群推断结果
括号中的数字表示每个人群对应族群成分的平均值。
用该体系对57个人群进行族群成分分析(图2),结果表明该体系可以对全球十大区域人群进行区分。当=3时,可以对亚洲、欧洲、非洲进行明确区分,维吾尔族(CUX)、东北亚的汉特(KTY)等混合人群的遗传成分呈现在欧洲和东亚族群成分的连续分布,当=4时,可以看出维吾尔族与汉特人群混合成分的差异,前者是欧洲(0.49)和东亚(0.44)成分的混合,而后者主要是欧洲(0.60)和美洲成分(0.31)的混合,这在实际应用中,有助于混合人群的进一步准确区分。随着值增加,先后在美洲、南亚、东南亚、北非、大洋洲、东北亚、西南亚出现新的族群成分,=10时,该体系可以对全球十大区域人群有较好的区分效力。地中海沿岸人群由于存在着广泛的基因交流,北非和西南亚人群当=10时才可以进行区分,并且南欧一些人群如由意大利和希腊人组成的TSG人群,有较多的西南亚成分。
通过图3可以找出人群特有的SNP位点,例如rs10108270, rs2986742, rs7238445和rs451722聚类在一起,且它们在南非人群中的频率明显高于其他人群,说明这些位点是南非人群特异。基因频率分布热图对所有人群的聚类结果与Structure分析=10时的结果基本相同,二者可以相互印证。
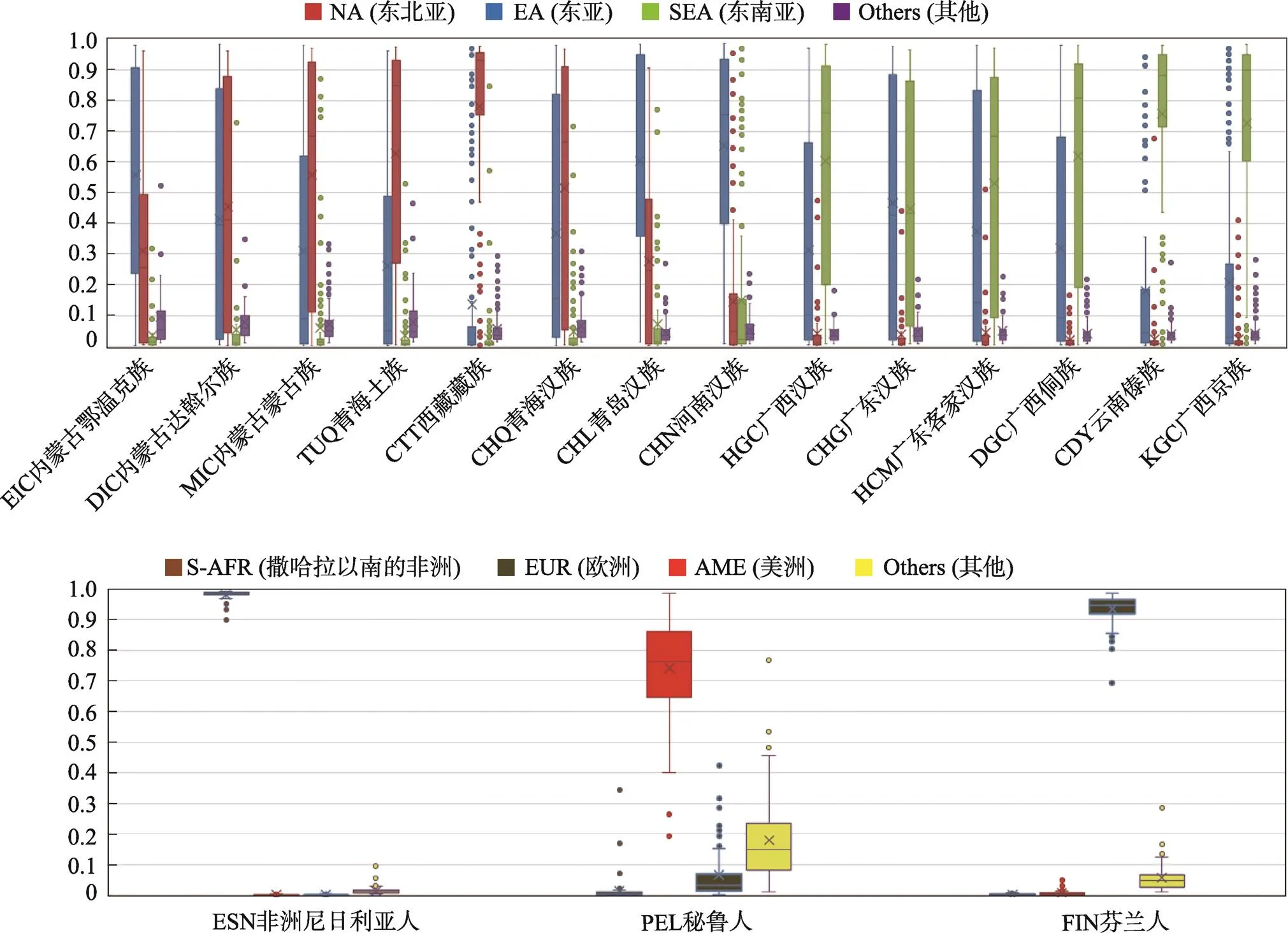
图4 基于17个测试人群1654个体的族群成分的箱线图
3.3 未知个体族群来源推断
本研究使用17个人群1654份个体作为测试数据集,计算其随机人群匹配概率、似然比和族群成分,结果见表3和图4。在所有测试样本中预测准确率较高的人群是遗传结构比较单纯的人群,如非洲尼日利亚人(ESN)的准确率为100%,欧洲芬兰人(FIN)准确率为100%,说明该体系对实际样本的区分能力较为稳定。
我国地处东亚,是一个多民族国家,中国南北方地区的一些少数民族人群在当地长期居住过程中形成了独特的体貌特征,随着战争、迁徙、通婚、融合等现象不断发生,不同人群之间出现基因交流,各地的汉族与当地的少数民族之间出现基因交流与融合,人群之间的差异是渐变的,中国地域人群的遗传结构复杂性在本研究测试人群中得到证实。
地处中国北方的达斡尔族(DIC)、鄂温克族(EIC)及蒙古族(MIC)人群是东胡后裔且都属于阿尔泰语系,在长期迁徙进化过程中与汉族人的基因交流等原因,部分个体被推断为东北亚人群或东亚和东北亚人群的混合[23,24](表3)。比如EIC-19号样本的AMP第一位人群为东北亚,与第二位人群的LR值大于10,该样本的东亚成分为0.55,东北亚成分为0.25,分析EIC-19号样本来源人遗传成分为东亚和东北亚人群混合。青海土族(TUQ)是鲜卑支系吐谷浑人后裔,在历史进程中不断吸收融合了羌、藏、汉、蒙古等民族的成分[25],本研究中,基于似然比统计TUQ的23名个体被推断为东北亚或者东亚和东北亚的混合(见表3),比如TUQ-71号样本,AMP第一位人群为东北亚,与第二位人群的LR值大于10,其东北亚成分为0.94,推断为东北亚,该结果与其历史起源相符。143名西藏藏族(CTT)个体中112名表现出大于0.7的东北亚遗传成分,其原因可能是藏缅语族人群的北方起源,杜若甫等[26]和Gayden等[27]对藏族常染色体遗传标记的研究证明了其北方起源。
汉族是中国的主体民族,源于北方古老的华夏部落[28,29],前期研究表明汉族人群具有混合特征,基于常染色体SNP频率的主成分分析呈现明显的南北分化[30]。图4A可以看出汉族人群自北向南表现出:北方成分逐渐减少,南方成分逐渐增多的趋势。表3的似然比统计结果中青海汉族(CHQ)、山东青岛汉族(CHL)和河南汉族(CHN)中国北方汉族人群的样本被推断为东亚人群的比例分别为74.0%、90.4%和81.3%,证明其对中国北方汉族人群推断的准确率较高。广西汉族(HGC)、广东客家汉族(HCM)和广东汉族(CHG)等中国南方汉族人群表现东亚和东南亚成分的混合,与自秦以来汉族人群的南迁及在迁徙过程中不断与南方少数民族交流融合等现象相符[31]。
广西京族(KGC)[32]约在16世纪初从越南的涂山等地迁来中国,和陆续迁来的汉族、壮族等各族人群进行了基因交流[33],广西侗族(DGC)和云南傣族(CDY)起源于南方的百越族[34,35]。在本研究中,KGC、DGC和CDY人群的族群成分以东南亚为主,混有一定比例的东亚成分(表3)。基于似然比的统计结果中部分个体被推断为东亚或者东亚和东南亚的混合,比如DGC-28号样本AMP第一位人群为东亚,与第二位人群的LR值大于10,其东亚成分为0.64,东南亚成分为0.17,族群推断为东亚和东南亚人群的混合,这可能与它们在历史进程中与汉族通婚、基因融合等有关。另外,民族是文化层面的概念,不同民族人群长期迁移与融合,族群推断结果可能出现与户籍登记不符的情况。在实际案件应用中,应综合分析似然比和族群成分。
综上所述,本研究前期基于质谱检测平台构建的74-plex SNPs复合检测体系在模板DNA量最低为1.5 ng时均可正确判型,适用于微量检材的检测。该体系实现了全球十个区域人群的区分,对东亚人群的南北方遗传成分可以进一步区分。检测结果可为案件提供更加详细的侦查线索。
[1] Tishkoff SA, Kidd KK. Implications of biogeography of human populations for 'race' and medicine., 2004, 36(11 Suppl): S21–S27.
[2] Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, Myers RM. Worldwide human relationships inferred from genome-wide patterns of variation., 2008, 319(5866): 1100–1104.
[3] Hellenthal G, Busby GBJ, Band G, Wilson JF, Capelli C, Falush D, Myers S. A genetic atlas of human admixture history., 2014, 343(6172): 747–751.
[4] Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, Jorde LB, Posukh OL, Sahakyan H, Watkins WS, Yepiskoposyan L, Abdullah MS, Bravi CM, Capelli C, Hervig T, Wee JT, Tyler-Smith C, van Driem G, Romero IG, Jha AR, Karachanak-Yankova S, Toncheva D, Comas D, Henn B, Kivisild T, Ruiz-Linares A, Sajantila A, Metspalu E, Parik J, Villems R, Starikovskaya EB, Ayodo G, Beall CM, Di Rienzo A, Hammer MF, Khusainova R, Khusnutdinova E, Klitz W, Winkler C, Labuda D, Metspalu M, Tishkoff SA, Dryomov S, Sukernik R, Patterson N, Reich D, Eichler EE. Global diversity, population stratification, and selection of human copy-number variation., 2015, 349(6253): aab3761.
[5] Phillips C. Forensic genetic analysis of bio-geographical ancestry., 2015, 18: 49–65.
[6] Santos C, Phillips C, Oldoni F, Amigo J, Fondevila M, Pereira R, Carracedo Á, Lareu MV. Completion of a worldwide reference panel of samples for an ancestry informative Indel assay., 2015, 17: 75–80.
[7] Halder I, Shriver M, Thomas M, Fernandez JR, Frudakis T. A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications., 2010, 29(5): 648–658.
[8] Kosoy R, Nassir R, Tian C, White PA, Butler LM, Silva G, Kittles R, Alarcon-Riquelme ME, Gregersen PK, Belmont JW, De La Vega FM, Seldin MF. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America., 2010, 30(1): 69–78.
[9] Nievergelt CM, Maihofer AX, Shekhtman T, Libiger O, Wang X, Kidd KK, Kidd JR. Inference of human continental origin and admixture proportions using a highly discriminative ancestry informative 41-SNP panel., 2013, 4(1): 13.
[10] Frudakis T, Venkateswarlu K, Thomas MJ, Gaskin Z, Ginjupalli S, Gunturi S, Ponnuswamy V, Natarajan S, Nachimuthu PK. A classifier for the SNP-based inference of ancestry., 2003, 48(4): 771–782.
[11] Phillips C, Salas A, Sánchez JJ, Fondevila M, Gómez-Tato A, Álvarez-Dios J, Calaza M, de Cal MC, Ballard D, Lareu MV, Carracedo A. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs., 2007, 1(3–4): 273–280.
[12] Wei YL, Wei L, Zhao L, Sun QF, Jiang L, Zhang T, Liu HB, Chen JG, Ye J, Hu L, Li CX. A single-tube 27-plex SNP assay for estimating individual ancestry and admixture from three continents., 2015, 130(1): 27–37.
[13] Wei L, Wei YL, Sun QF, Wang YY, Li CX. The development of a 27-plex SNP multiplex system., 2016, 31(1): 13–17.魏丽, 魏以梁, 江丽, 孙启凡, 王英元, 李彩霞. 27-plex SNPs复合扩增检测体系构建与应用评价. 中国法医学杂志, 2016, 31(1): 13–17.
[14] Jiang L, Sun QF, Ma Q, Zhao WT, Liu J, Zhao L, Ji AQ, Li CX. Optimization and validation of analysis method based on 27-plex SNP panel for ancestry inference., 2017, 39(2): 166–173.江丽, 孙启凡, 马泉, 赵雯婷, 刘京, 赵蕾, 季安全, 李彩霞. 27-plex SNP种族推断方法的优化及验证. 遗传, 2017, 39(2): 166–173.
[15] Li CX, Pakstis AJ, Jiang L, Wei YL, Sun QF, Wu H, Bulbul O, Wang P, Kang LL, Kidd JR, Kidd KK. A panel of 74 AISNPs: Improved ancestry inference within Eastern Asia., 2016, 23: 101–110.
[16] Ma M, Liu J, Hu S, Zhang T, Zhou H, Feng BQ, Liu HB, Li B, Li CX. The validation study of 74-plex SNP assay for ancestry inference., 2019, 34(4): 324–329.马咪, 刘京, 胡胜, 张涛, 周浩, 冯保强, 刘海渤, 李蓓, 李彩霞. 74重SNP族群来源推断体系准确性验证研究. 中国法医学杂志, 2019, 34(4): 324–329.
[17] Ren P, Liu J, Zhao H, Fan XP, Xu YC, Li CX. Construction of a rapid microfluidic-based SNP genotyping (MSG) chip for ancestry inference., 2019, 41: 145–151.
[18] Clendenen TV, Rendleman J, Ge W, Koenig KL, Wirgin I, Currie D, Shore RE, Kirchhoff T, Zeleniuch-Jacquotte A. Genotyping of single nucleotide polymorphisms in DNA isolated from serum using sequenom MassARRAY technology., 2015, 10(8): e0135943.
[19] Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles., 2007, 7(4): 574–578.
[20] Liu J, Li S, Jang L, Zhao L, Zhao WT, Feng L, Liu HB, Ji AQ, Li CX. DNA Ancestry Analyzer: an automatic program for ancestry inference of unknown individuals., 2018, 22(1): 3–7, 41.刘京, 李盛, 江丽, 赵蕾, 赵雯婷, 丰蕾, 刘海渤, 季安全, 李彩霞. 对于未知来源个体进行族群推断的自动分析系统. 生命科学研究, 2018, 22(1): 3–7, 41.
[21] Phillips C, McNevin D, Kidd KK, Lagacé R, Wootton S, de la Puente M, Freire-Aradas A, Mosquera-Miguel A, Eduardoff M, Gross T, Dagostino L, Power D, Olson S, Hashiyada M, Oz C, Parson W, Schneider PM, Lareu MV, Daniel R. MAPlex - A massively parallel sequencing ancestry analysis multiplex for Asia-Pacific populations., 2019, 42: 213–226.
[22] Sun K, Yun LB, Zhang C, Shao CC, Gao TZ, Zhao ZQ, Hou YP, Xie JH, Tang QQ. Evaluation of 12 Multi-InDel markers for forensic ancestry prediction in Asian populations., 2019, 43: 102155.
[23] Xu Y, Zhang XL, Zhang QC, Cui YQ, Zhou H, Zhu H. Genetic relationship between ancient Khitan and modern Daur., 2006, 44(6): 997–1000.许月, 张小雷, 张全超, 崔银秋, 周慧, 朱泓. 古代契丹与现代达斡尔遗传关系分析. 吉林大学学报(理学版), 2006, 44(6): 997–1000.
[24] Zhu H. The ethnic type and related issues of the Khitay., 1991(2): 36–41.朱泓. 契丹族的人种类型及其相关问题. 内蒙古大学学报(哲学社会科学版), 1991, (2): 36–41.
[25] Fan H. Relationship among 28 Chinese populations in western and southern of China based on STR loci., 2006.范浩. 应用STR位点研究中国西、南部28个民族群体族源关系. 昆明医学院, 2006.
[26] Du R, Xiao C, Cavalli-Sforza LL. Genetic distances between Chinese populations calculated on gene frequencies of 38 loci., 1997, 40(6): 613–621.
[27] Gayden T, Mirabal S, Cadenas AM, Lacau H, Simms TM, Morlote D, Chennakrishnaiah S, Herrera RJ. Genetic insights into the origins of Tibeto-Burman populations in the Himalayas., 2009, 54(4): 216–223.
[28] Zhao YB, Yu CC, Zhou H. Study on the origin and development of the Han Chinese., 2012, 33(4): 45–49.赵永斌, 于长春, 周慧. 汉族起源与发展的遗传学探索. 吉林师范大学学报(自然科学版), 2012, 33(4): 45–49.
[29] Huang YZ. The historical migration of the Han population and the color pattern of the southern Han folk songs, 1989(4): 36–48.黄允箴. 汉族人口的历史迁徙与南方汉族民歌的色彩格局. 中国音乐学, 1989, (4): 36–48.
[30] Xu SH, Yin XY, Li LS, Jin WF, Lou HY, Yang L, Gong XH, Wang HY, Shen YP, Pan XD, He YG, Yang YJ, Wang Y, Fu WQ, An Y, Wang JC, Tan JZ, Qian J, Chen XL, Zhang X, Sun YF, Zhang XJ, Wu BL, Jin L. Genomic dissection of population substructure of Han Chinese and its implication in association studies., 2009, 85(6): 762–774.
[31] Wang YS. Historical evolution of the Han population and its survival area., 2010, (6): 3–7.王跃生. 汉族人口及其生存区域的历史演变. 历史教学(下半月刊), 2010, (6): 3–7.
[32] Mo L, Wang CL. Analysis on the characteristics of Guangxi Jing population., 1990, (3): 17–22.莫龙, 王春林. 广西京族人口特点浅析. 广西民族研究, 1990, (3): 17–22.
[33] Jin TB, Gao Y, Chen T, Yan HX, Li SB. Genetic relationships of 15 populations of Guangxi province., 2004, 25(5): 422–424, 429.金天博, 高雅, 陈腾, 阎春霞, 李生斌. 广西地区15个不同民族人群的群体遗传学关系. 西安交通大学学报 (医学版), 2004, 25(5): 422–424, 429.
[34] Tang JP, Yu X, Jiang FH, Yu XJ. Analyzing population differentiation between Hall and other population of Guangxi., 2008, 31(6): 409–412.唐剑频, 于昕, 蒋丰慧, 于晓军. 广西汉族群体与其他群体的群体差异分析. 国际遗传学杂志, 2008, 31(6): 409–412.
[35] Qian YP. Rearch on genetic diversity of five Yunnan ethnic groups in China[Dissertation]., 1999.钱亚屏, 中国云南5个民族的遗传多样性研究[学位论文]. 中国协和医科大学, 1999.
The ancestry inference of Chinese populations using 74-plex SNPs system
Yang Liu1,2, Changchun Sun1,2, Mi Ma2,3, Ling Wang2, Wenting Zhao2, Quan Ma2, Anquan Ji2, Jing Liu2, Caixia Li1,2
A panel of ancestry informative SNPs (AISNPs) can be used to analyze the genetic components of a population and infer the ancestral origin of a DNA sample. Previously, we have selected a 74-AISNPs panel and used it to infer the ancestry of unknown individuals in the following ten geographical regions: Sub-Saharan Africa, North Africa, Europe, Pacific, Americas, Southwest Asia, South Asia, North Asia, East Asia and Southeast Asia. We have also established a 74-plex SNPs assay based on SEQUENOM system. In the present study, we genotyped 1371 individuals from 14 populations of China using this multiplex assay, and validated its ability to infer the ancestry in Chinese populations. Firstly, based on the reference database of 3628 individuals from 57 world populations, Structure and Heatmap were employed to evaluate the population differentiation capacity. The training data include 1654 individuals from 14 Chinese populations and 3 populations from 1K Genome, which are not included in the reference database. Then the likelihood ratio and ancestry components were analyzed for individual ancestry assignment using the 74-plex SNPs. The minimum amount of DNA required for a full genotype of the 74 SNPs is 1.5 ng, which is applicable for forensic analysis. The results demonstrate that this system can be used in differentiating the population from ten geographical regions. The ancestry inference accuracy for EUR/SAFR/AME population is 95.4%, 71.0% for East Asia and 66.4% for Southeast Asia respectively. The ancestry inference inclusive rate for EUR/SAFR/AME population is 1.06%, 17.9% for East Asia and 33.3% for Southeast Asia respectively. The results suggest that this method can be used in forensic investigations of criminal cases.
forensic genetics; AISNPs; ancestry inference; Chinese populations
2019-11-13;
2020-01-13
国家自然科学基金项目(编号:81772027),国家重点研发计划资(编号:2017YFC0803501),国家科技资源共享服务平台计划项目(编号:YCZYPT[2017]01-3)和中央公益类基本科研业务费专项资金项目(编号:2017JB025,2017JB026,2019JB011)资助[Supported by the National Natural Science Foundation of China (No.81772027), National Key R&D Program of China (No. 2017YFC0803501), National Science and Technology Resources Sharing Service Platform Project (No. YCZYPT[2017]01-3) and Fundamental Research Funds for Institute of Forensic Science (Nos. 2017JB025, 2017JB026, 2019JB011)]
刘杨,硕士研究生,研究方向:法医学。E-mail: 1979227825@qq.com
李彩霞,博士,主任法医师,研究方向:法医遗传学。E-mail: licaixia@tsinghua.org.cn
刘京,硕士,主检法医师,研究方向:法医遗传学。E-mail: biojing@yeah.net
10.16288/j.yczz.19-252
2020/2/10 16:00:49
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20200210.1533.002.html
(责任编委: 赖江华)
猜你喜欢
金桥(2022年6期)2022-06-20
英美文学研究论丛(2021年2期)2021-02-16
国际汉学(2020年1期)2020-05-21
时代邮刊(2019年24期)2019-12-17
船舶标准化工程师(2019年4期)2019-11-29
知识经济·中国直销(2018年9期)2018-09-28
初中生世界·七年级(2017年7期)2017-09-04
东北史地(学问)(2017年1期)2017-06-15
东北史地(学问)(2017年1期)2017-06-15
通化师范学院学报(2016年11期)2017-01-15
