
肝细胞癌全基因组关联分析研究进展及展望
2020-03-28 10:58周泽文张若昕魏庆义余红平
中国癌症防治杂志 2020年1期
周泽文张若昕魏庆义余红平
作者单位:530021南宁 1广西医科大学附属肿瘤医院;2广西医科大学公共卫生学院;200433上海 3复旦大学公共卫生学院流行病学教研室;4教育部公共安全重点实验室;5复旦大学附属肿瘤医院肿瘤研究所
2018年全球肿瘤统计分析结果显示[1],全球新发癌症病例数为18 078 957例,其中肝癌新发病例841 080例,占全球肿瘤新发病例的4.65%,位列第六;死亡病例9 555 027例,其中肝癌死亡病例数为781 631例,占全球肿瘤死亡病例的8.18%,位列第四。肝癌是世界范围内常见的高发肿瘤,严重威胁人类健康。肝癌发病率在世界范围内差异很大,在东亚、东南亚和撒哈拉以南非洲最高,而全球肝癌患者中约有一半在中国[2]。我国的肝癌类型与欧美和日本等国家不同,乙型肝炎病毒(hepatitis B virus,HBV)感染是我国肝癌发生的最主要原因[3]。肝癌恶性程度高、病程进展迅速、易复发和转移,确诊患者多以中、晚期为主,5年生存率仅为4%左右,仍是目前临床上治疗效果最差的肿瘤之一[4-5]。虽然超过80%的肝癌与慢性乙型病毒性肝炎相关,但最终只有一小部分乙型病毒性肝炎患者罹患肝癌,提示不同个体对肝癌存在遗传易感性,遗传因素在肝癌的发生中起重要作用[6-8]。阐明肝癌的易感基因,不仅有助于深入理解肝癌的发病机制,而且能为预测个体肝癌发生风险、早期预防、个体化治疗及新型高效药物的筛选提供理论依据和生物靶标,具有重要意义。
全基因组关联分析(genome-wide association study,GWAS)研究是在全基因组层面上开展的多中心、大样本、多阶段验证的遗传变异,即单核苷酸多态性(single nucleotide polymorphism,SNP)与疾病的关联性研究,使我们可能在全基因组范围内发现与疾病相关的易感基因及其遗传变异[9]。GWAS已被科学界公认是行之有效的系统搜寻重大疾病易感基因的研究方法,并且这一方法无需预先提出致病基因的假设,这一点与候选基因策略不同,已经成为研究人类肿瘤遗传变异的有力工具[10-13]。GWAS的流程示意图如图1所示。自2010年第一篇关于肝细胞癌(hepatocellular carcinoma,HCC)的GWAS文章[14]发表以来,有关HCC的GWAS研究已有20余篇。本文将对目前GWAS在HCC中的研究进行总结,并分析GWAS在HCC研究中的优势、局限性和未来展望。
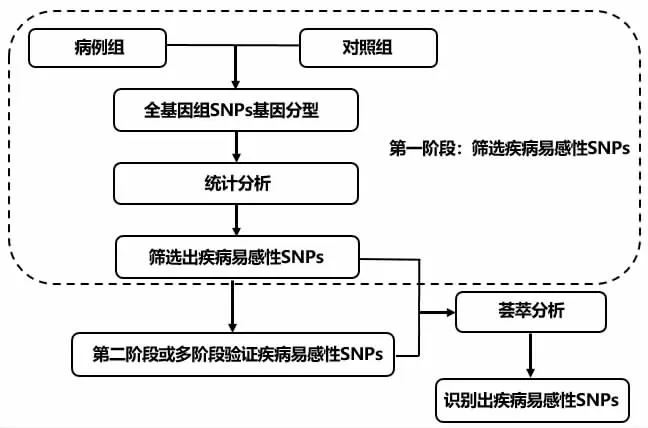
图1 GWAS流程示意图
1 GWAS研究的HCC人群
目前关于HCC的GWAS研究集中在东亚人群,研究主要采用两阶段法或多阶段法,第一阶段在覆盖全基因组范围的SNP进行关联性分析,筛选出与HCC遗传易感性相关联的SNP位点,接着对这些关联的SNP进行第二阶段或多阶段的大样本量人群进一步验证,然后结合两阶段或多阶段结果进行综合分析。HCCGWAS研究中的病例组为HCC患者,对照组为慢性乙型病毒性肝炎患者、慢性丙型病毒性肝炎患者或肝硬化患者,涉及的研究人群主要来自中国、日本、韩国和泰国。
2010年,张红星等首次报道了我国HBV相关性HCC GWAS研究,并确定1p36.22为HCC的易感区域[14]。同年,CLIFFORD等[15]报道了韩国人群HCC GWAS研究结果,发现了与拷贝数变异显著关联的T细胞受体γ和α基因易感区域和包括MHC-Ⅱ区域在内的3个遗传位点。2011年,KUMAR等[16]开展了日本丙型肝炎病毒(hepatitis C virus,HCV)相关性HCC GWAS研究,发现位于基因座6p21.33区域的SNP rs2596542位点与HCV相关性HCC易感性显著相关。文献[14-25]报道的GWAS研究发现的HCC易感区域和位点详见表1。
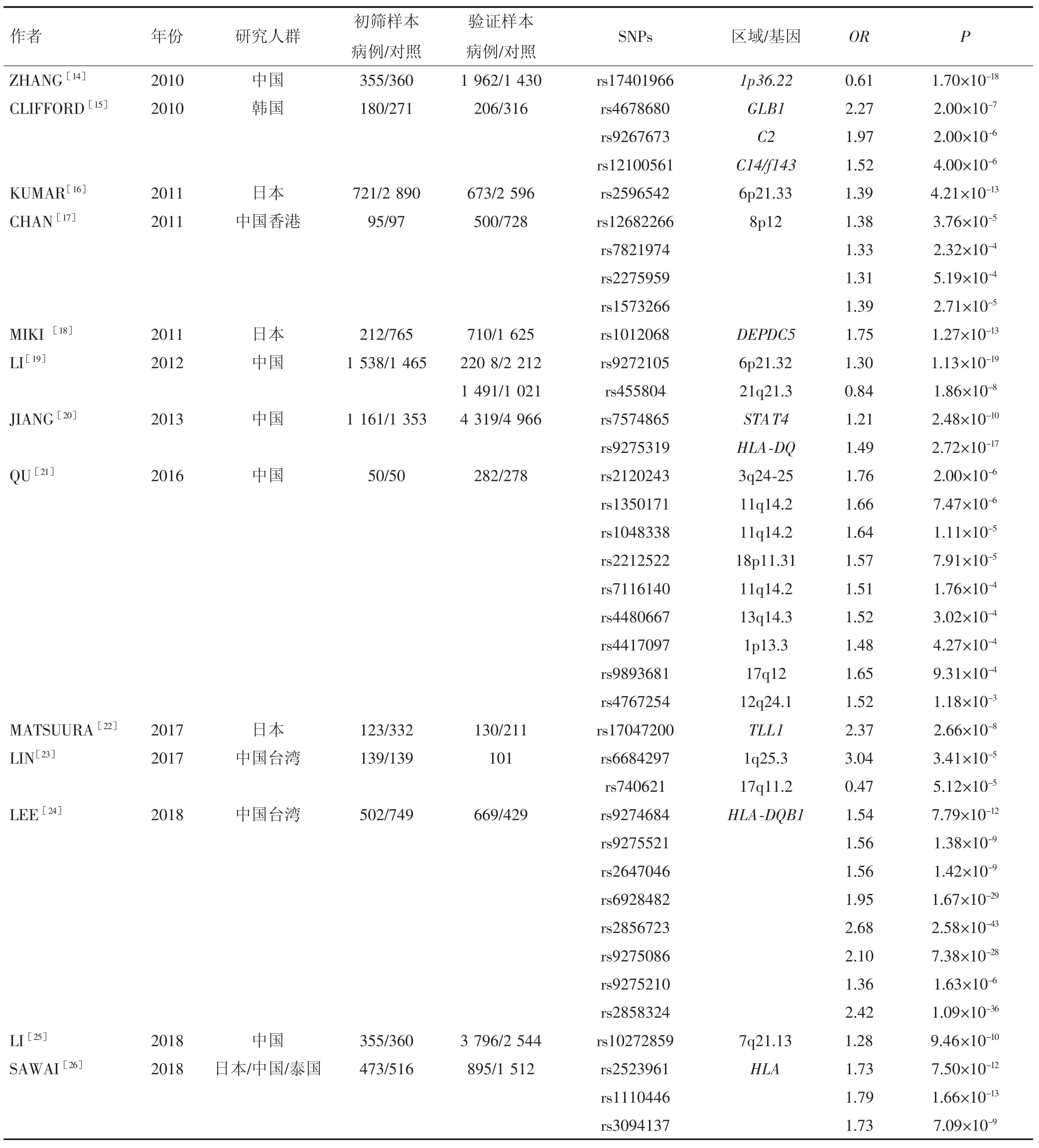
表1 GWAS研究发现的HCC易感区域和位点
2 GWAS发现的HCC易感区域和位点
2.1 染色体1p36.22区域
1p36.22区域是最早被发现与HCC遗传易感性相关的区域,在中国人群开展的一项GWAS研究[14]在该区域发现了1个SNP位点(KIF1β基因的内含子SNP:rs17401966)和2个可能的致病基因UBE4β和PDG,表明1p36.22区域是HBV相关性HCC的遗传易感区域,这一区域KIF1β,UBE4β与PGD基因相关的表达产物或生物学通路途径可能参与了HCC的发生发展过程。后续的研究[27]显示,KIF1β是HCC的抑癌基因,该基因通过整联蛋白通路调控细胞侵袭、迁移和转移,其低表达与HCC的高转移风险显著相关。
2.2 染色体6p21.3区域
染色体6p21.3区域同时存在HBV相关性和HCV相关性HCC的遗传易感位点。前期的肝癌GWAS研究结果显示[28-29],SNP位点变异与宿主感染HCV后发展成慢性丙型病毒性肝炎密切相关,但是否与HCV相关性HCC相关联尚缺乏研究证据。为了确定HCV相关性HCC遗传易感位点,一项GWAS研究[16]在日本人群中开展,结果发现位于染色体6p21.33区域的SNP位点rs2596542与HCV易感性无关联(P=0.61),但在慢性丙型病毒性肝炎发展为HCC的进程中发挥重要作用。进一步研究结果显示,该SNP位点rs2596542位于主要组织相容性复合体的Ⅰ类相关链A(major histocompatibility complex classⅠ-related chain A,MICA)基因座,并且可以影响该基因的表达,携带rs2596542A等位基因的HCC患者中MICA的表达呈低水平。前期有研究结果[30]显示,血清可溶性MICA水平与膜结合的MICA25水平成正比,而与膜结合的MICA25对激活自然杀伤细胞和CD8+T细胞以消除感染了HCV的细胞至关重要,这一系列的研究表明,宿主免疫系统遗传变异在HCV相关性HCC发生中起关键作用。
前期GWAS研究[31-32]表明,人类白细胞相关抗原(human leukocyte antigen,HLA)编码区域的变异与HBV感染之间存在关联性,HLA-DRB1位点的遗传变异与持续感染HBV的风险密切相关。一项在中国开展的HBV相关性HCC的GWAS研究[19]发现,染色体6p21.32区域的SNP位点rs9272105与HBV相关性HCC易感性有关联,该位点位于6p21.32区域的HLA-DQA1和HLA-DRB1之间。在中国开展的另一项GWAS研究[20]对在日本慢性丙型病毒性肝炎人群发展成HCC起到关键作用,位于染色体6p21.33区域的SNP位点rs2596542在中国HBV相关肝癌人群中进行了检验,发现该位点与HBV相关性HCC不相关(OR=0.93,P=0.24)。这种关联的差异提示HCV和HBV相关性HCC易感性的不同遗传背景。一项在台湾地区开展的HCV相关性HCC GWAS研究也在染色体6p21.3的HLA区域发现了HCC相关的8个易感位点,这8个易感位点均位于HLA-DQB1基因座中,而且HCV基因型与HLA-DQB1之间存在HCC风险的相互作用[24]。
2.3 染色体7q21.13区域
一项在中国HBV相关性HCC人群开展的GWAS研究发现染色体7q21.13区域存在HCC的新关联位点rs10272859,该位点位于CDK14基因内的第5个内含子中。前期研究表明,CDK14是细胞周期蛋白依赖性激酶(cyclin-dependent kinases,CDK)家族成员,该家族在细胞周期进程和细胞增殖中起关键作用[33]。CDK14的移位表达可诱导HCC细胞大量侵袭和迁移[34]。此外,CDK14表达增加与低分化程度HCC以及微血管浸润有关[35]。该项GWAS研究进一步证实了CDK14表达上调与肝癌患者总生存期减少有关,且SNP位点rs10272859参与调控CDK14表达并影响HCC患者预后[25]。
2.4 染色体21q21.3区域
抑制谷氨酸释放和(或)谷氨酸受体活性可以抑制乳腺癌[36]、喉癌[37]、胰腺癌[38]和胃癌[39]肿瘤细胞增殖和侵袭,表明谷氨酸及其信号通路在多种癌症发展中起重要作用。在中国人群中开展的一项GWAS研究[19]将染色体21q21.3区域的SNP位点rs455804鉴定为HCC的易感位点。rs455804位于GRIK1基因的第1个内含子中,而GRIK1编码参与谷氨酸信号传导的信号蛋白CLUR5,这项研究不仅表明了谷氨酸信号通路在肝癌发展过程中的潜在作用,也为谷氨酸信号通路在癌症发展中的重要作用提供了新的证据。
2.5 STAT4基因座
STAT4传输白介素12和Ⅰ型干扰素(IFN-α或IFN-β)的信号,以诱导IFN-γ产生[40]。IFN-γ是一种多效细胞因子,在宿主防御中起着至关重要的作用[41]。STAT4对IFN-γ的激活受损可能会降低其抗病毒和抗肿瘤活性,而IFN-γ过表达则可能导致自身免疫性疾病的发展[42-43]。此前,多个GWAS和基于候选基因的关联研究报告了STAT4遗传变异,特别是SNP位点rs7574865与各种自身免疫性疾病发病风险有关联[44-45]。一项HBV相关性HCC的GWAS研究发现SNP位点rs7574865的G等位基因与HCC发生风险较高相关[20],而该等位基因与自身免疫疾病发生风险较低相关,这一结果支持STAT4在自身免疫性疾病和HBV相关性HCC发病中的双重作用。
2.6 其他区域
GWAS研究发现的其他易感位点所在染色体区域主要有1p13.3、1q25.3、3q24、8p12、11q14.2、12q24.1、13q14.3、17q11.2、17q12和18p11.31等,这些区域的位点或位于基因荒漠区,或所在区域基因功能尚未完全阐明,故目前对这些区域位点的功能研究较少,尚待后续研究进一步探索阐明其在HCC发生发展过程中的机制作用。
3 GWAS在HCC研究中的其他应用
3.1 化学免疫治疗机制研究
MICA基因为HCV相关性HCC的易感基因,在HCC患者中MICA蛋白往往呈低表达[16]。鉴于MICA蛋白在免疫激活中的重要作用[46],调节MICA表达的药物可能是HCC免疫治疗的新靶标。基于上述结果,GOTO等[47]通过删选在FDA批准的药物库中发现组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor,HDACi)可以恢复MICA表达,进一步的代谢组学分析结果显示,HDACi诱导的HCC细胞特异性MICA表达增强可以增强自然杀伤细胞介导的细胞毒性,MICA脱氢酶抑制剂处理可以进一步增强这种毒性,两者都增强了自然杀伤细胞的抗肿瘤活性,进而起到抑制肿瘤生长的作用,这一研究结果揭示了肿瘤免疫治疗的新机制,为HCC化学免疫治疗提供新思路。
3.2 HCC患者术后生存时间预测模型
GWAS已在STAT4和HLA-DQ以及HLA I类区域的基因座和1p36.22、6p21.32、7q21.13及21q21.3等处鉴定了HCC的遗传易感位点[14-26]。但是,与HCC患者术后生存相关的基因位点知之甚少。由于HCC病情复杂,单个基因变异不可能对HCC的临床结果产生巨大影响,但是一系列SNP的协同可能对疾病进展起重要作用[48-49]。近期在中国开展的一项以HCC术后患者为研究对象的GWAS研究,研究第一阶段通过367例HCC患者鉴定与预后相关的SNPs,第二阶段在758例患者中对发现的与预后相关的SNP位点进行验证,研究最终确定了5个SNP位点(rs10893585、rs2431、rs34675408、rs6078460和rs6766361)与患者生存时间显著相关,随后通过Cox比例风险回归建立预后模型预测HCC患者术后生存,生存预测模型的敏感性和特异性分别为58.8%和74.8%,用该模型计算HCC患者术后5年生存率准确可靠[50]。
3.3 基因型-表型关联分析及易感基因与SNP的功能机制研究
目前,GWAS研究鉴定出了大量HCC易感区域和基因,基于这些易感基因开展基因型-表型及相关分子作用途径的研究成为新的研究热点。大量研究证据表明TP53基因在HCC发生发展中起重要作用[51]。有研究者基于其前期一项GWAS研究探讨HBV相关性HCC患者中与TP53蛋白表达状态相关的候选SNPs和分子通路,阐明其可能的机制,并提出SNP-togene-to-pathway的科学假说。该研究发现了18个与HBV相关性HCC中TP53表达状态相关的候选SNPs和10个候选通路,其中最强的作用机制涉及主要组织相容性复合体Ⅱ类DP beta1(HLA-DPB1-rs1042153)、主要组织相容性复合体Ⅱ类DQ beta 1(HLA-DQB1-rs1130399、HLA-DQB1-rs1049056、HLA-DQB1-rs1049059和HLA-DQB1-rs1049060)和主要组织相容性复合体Ⅱ类DR beta 1(HLA-DRB1-rs35445101),在此基础上提出了5个新的SNP-to-gene-to-pathway假说;生存分析表明,TP53阴性状态下的COL6A3-rs111231885、COL6A3-rs113155945和COL6A3区域的4 CC单倍型可能对肝切除术后HBV相关的HCC患者具有保护作用,并且可以作为潜在的预后评估生物标志物[52]。以往研究结果显示,MKI67标记指数与包括肝癌在内的多种肿瘤的临床病理特征和临床预后相关[53-54],而NM23在肿瘤组织中的表达与HCC患者肿瘤转移和生存期有关[55]。两项分别针对MKI67和NM23蛋白表达与HCC患者预后的GWAS研究在中国开展,针对MKI67蛋白表达的GWAS研究结果发现,靠近TNN和CCDC8的5个位点(rs3813243、rs2288563、rs2562832、rs56142888和rs34186470)与MKI67蛋白表达有关,位于TTN基因SNP位点rs2288563和rs25622832可以作为HBV相关性HCC患者的临床预后标志物[56];而针对与NM23蛋白表达水平相关的候选SNP研究结果显示,银屑病易感性1候选物1(PSORS1C1)和含StAR相关脂质反式结构域3(STARD3)的SNP与NM23表达相关,PSORS1C1的遗传变异是预测HBV相关性HCC患者术后临床结局的潜在生物标志物,PSORS1C1和STARD3的遗传变异与HBV相关性HCC患者NM23表达和临床结局有关[57]。
4 GWAS在HCC研究中的优势、局限性和展望
4.1 GWAS的优势
GWAS在研究人群基因组中对数十万至数百万个遗传变异进行分析以鉴定基因型-表型关联,在过去十年间已经彻底改变了复杂疾病的遗传学研究方式[58-59]。在GWAS之前,为了研究HCC易感性与SNP的关联,研究者主要采用候选基因策略[60-61]。该策略着眼于预设的候选基因而遗漏了基因组内大部分的遗传信息。GWAS研究的最大优势在于摒弃了候选基因方法中的预先假设,不再着眼于已知的生物学通路基因,而是从人类全基因组范围内筛选出与复杂疾病易感性关联的遗传变异[62-63]。此外,GWAS研究一般样本量较候选基因方法大,采用更严格的检验水准,且往往进行多阶段、多中心验证,研究结果的可靠性得到了有效保障。
4.2 GWAS存在的问题
GWAS研究也存在局限性。首先,HCC GWAS研究往往基于严格的检验水准[64],一般只选择少量的峰值位点进行后期验证,降低了假阳性,但同时也可能损失其他潜在的遗传位点;其次,目前大部分的研究主要聚焦在SNP位点信息与遗传易感性的关联性,一定程度上忽略了拷贝数变异(copy number variations,CNVs)、基因缺失、串联重复序列等其他变异,而且对基因-环境因素交互作用也缺乏足够重视[65];再次,目前GWAS研究主要在于发现关联性强的SNP位点,这些位点大多位于基因的非编码区或基因和基因之间的结构区域甚至基因的荒漠区[66],对于这些SNP的生物学功能仍处于探索阶段,这也极大限制了GWAS研究结果的实际应用;最后,不可回避的问题是任何肝癌GWAS研究中鉴定的单核苷酸变异只能解释复杂性状的遗传性的一小部分[67-68],而且关联位点和肝癌发生发展并不一定存在因果关系。对于肝癌这一复杂多基因疾病,寻找全面的致病遗传标志物和基因仍是未来的重要研究方向。
4.3 GWAS未来展望
GWAS改变了HCC的遗传学研究,已经开展的GWAS研究不仅阐述了HCC发生、发展的遗传学基础,积累了丰富的遗传学数据,也为检测、治疗、预后评估以及药物开发提供了宝贵的遗传生物标志物信息。随着新的分子生物学技术与新方法的出现以及基于数据挖掘的生物信息学研究方法研究的不断深入,后GWAS时代已经来临。基于HCC GWAS发现的SNP以及在同一区域中连锁的SNP位点可多达成百上千个,如何从中找到真正与HCC相关联的SNP,并从生物学上解释其功能及其与HCC的关系,是后GWAS时代面临的重大挑战,也是今后复杂疾病GWAS领域研究的方向和热点。对HCC GWAS鉴定出的疾病易感基因功能研究的不断深入,将有助于人类明确认知基因变化与HCC的关系,从分子水平层面诠释HCC的发病机制,发现新的HCC相关标志物和诊疗靶点,改善HCC的预防、诊断与治疗,促进精准医学理念下HCC预防与治疗的发展,进一步提升我国HCC的综合防治水平。
猜你喜欢
分子催化(2022年1期)2022-11-02
心理学报(2022年2期)2022-02-15
园林科技(2021年2期)2022-01-19
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
电子制作(2019年24期)2019-02-23
电脑知识与技术(2018年19期)2018-11-01
现代仪器与医疗(2017年2期)2017-05-10
中国现代医生(2014年23期)2014-09-02
