
枯草芽胞表面CotC和CotG共展示海藻糖合酶研究
2020-02-29 08:49刘洪玲王腾飞
天津科技大学学报 2020年1期
刘洪玲,刘 浩,王腾飞
(1. 工业发酵微生物教育部重点实验室,天津科技大学生物工程学院,天津300457;2. 齐鲁工业大学(山东省科学院)生物工程学院,济南250353)
芽胞是有某些细菌产生的坚韧且不具有繁殖能力的结构,可以使这些细菌在不利的条件下处于休眠状态.芽胞的结构由外到内依次为芽胞外壁、芽胞衣壳、芽胞皮层和芽胞髓质.芽胞髓质由芽胞壁、芽胞质膜、芽胞质和染色体核心这四部分组成的[1].芽胞的形成是一系列调控基因和结构基因按一定时序调控表达的过程,这些基因包括芽胞衣壳蛋白基因以及调控基因,如 cotA、cotB、cotC、cotF、cotG 等 20余种[2].芽胞衣壳是一种由至少 70种蛋白组成的复杂结构[3].芽胞外壁从结构上又可详细分为 Crust层、外层、内层和基底层[4-5].虽然芽胞对热、辐射、静水压、极端 pH环境和一些有毒化学物品等有很强的抵抗能力[6],但当芽胞遇到合适的萌发条件时却极为敏感,芽胞会迅速进入萌发状态,迅速出芽生长,恢复到营养细胞的状态[7].芽胞表面展示技术,是一项在芽胞表面展示具有生物活性外源蛋白的技术.该技术将外源蛋白基因与芽胞衣壳蛋白基因进行融合,进而将外源蛋白和锚定蛋白共同展示于芽胞表面.Isticato等[4]首次成功实现了枯草芽胞杆菌芽胞衣壳蛋白融合展示外源蛋白的表达,此后芽胞表面展示技术受到了广泛的关注.枯草芽胞表面展示技术应用最普遍的原因主要是芽胞具有结构清楚[8-9]、基因改造简单[10]、全基因组数据已公布[11]等优点.枯草芽胞表面的衣壳蛋白分子质量较大,这使得芽胞表面展示系统能够展示分子质量较大的蛋白,而且由于芽胞衣壳蛋白是内源性蛋白,所以在引导外源蛋白展示的过程中,能够大幅度地提高外源蛋白的折叠效率和展示效率[2].Mauriello等[5]通过将携带外源蛋白基因的锚定蛋白CotC和枯草芽胞杆菌基因组中淀粉酶基因进行同源重组的方式,成功地在芽胞表面展示了破伤风毒素(TTFC)和埃希氏大肠杆菌嗜热毒素(LTB).Isticato等[4]利用CotB作为芽胞表面展示外源抗原的载体蛋白,将破伤风毒素(TTFC)C末端的 459个氨基酸片段展示在枯草芽胞杆菌芽胞表面.Kwon等[12]以 CotG为蛋白载体将半乳糖苷酶展示于枯草芽胞杆菌芽胞表面,重组芽胞在水–有机相反应体系中具有半乳糖苷酶催化活性,并且较化学方法固定于芽胞表面的半乳糖苷酶有更好的稳定性.Nguyen等[13]也利用锚定蛋白 CotB、CotC和 CotG在芽胞表面成功展示了GFPuv蛋白,并利用CotB自身的启动子和替换后的 IPTG诱导启动子,实现了 GFPuv蛋白的调控表达.
本文拟以CotC、CotG作为TreS的锚定蛋白,构建整合型表达质粒,将 TreS展示于芽胞表面.通过芽胞表面展示技术,将 TreS展示于枯草芽胞杆菌表面,形成微固定化酶,实现 TreS在芽胞表面的展示表达.
1 材料与方法
1.1 材料
1.1.1 菌株、质粒与培养基
大肠杆菌(Escherichia coli)DH5α,由本实验室保存;枯草芽胞杆菌(Bacillus subtilis)WB800n,德国MoBiTec公司;pDG1730,华中农业大学;pPIC9k、pPIZαA,杭州宝赛生物科技有限公司.
DSM 培养基:营养肉汤 8g/L,KCl 1g/L,MgSO4·7H2O 0.25g/L,121℃高压灭菌 20min 后加入无菌的Ca(NO3)21mmol/L、MnCl20.01mmol/L和FeSO40.01mmol/L.
LB培养基:蛋白胨10g/L,酵母浸粉5g/L,NaCl 10g/L.在液体培养基中添加 20g/L的琼脂,即为固体培养基.
GM 培养基:蛋白胨 10g/L,酵母浸粉 5g/L,NaCl 10g/L,山梨醇 0.5mol/L,用去离子水定容至1L.
ETM 培养基:山梨醇 0.5mol/L,甘露醇0.5mol/L,甘油10%,用去离子水定容至1L.
RM 培养基:蛋白胨 10g/L,酵母浸粉 5g/L,NaCl 10g/L,山梨醇0.5mol/L,甘露醇0.38mol/L,用去离子水定容至1L.
TB培养基:胰蛋白胨 12g/L,酵母提取物24g/L,甘油 4mL/L,K2HPO472mmol/L,KH2PO417mmol/L.
1.1.2 主要试剂
壮观霉素,北京吉美生物技术有限公司;氨苄青霉素,上海慕远生物科技有限公司;氯霉素、细菌DNA小量提取纯化试剂盒、SanPrep柱式DNA胶回收试剂盒、Ezup柱式细菌基因组 DNA抽提试剂盒、蛋白 marker,上海生工生物工程有限公司;高纯度质粒小提试剂盒、多片段无缝克隆试剂盒、核酸DL5000marker、TaqDNA 聚合酶、2×Phanta Max Master Mix,南京诺唯赞生物科技有限公司;限制性内切酶、T4DNA 连接酶,赛默飞世尔科技(中国)公司;溶菌酶,NOVO 公司;电泳级琼脂糖,西班牙Biowest Agarose 公司;10×Loading Buffer,日本Takara公司;6×His、His-Tag鼠单克隆抗体,美国Proteintech公司;BSA、山羊血清、BCA 蛋白浓度测定试剂盒、FITC–羊抗小鼠 IgG,武汉博士德生物工程有限公司;辣根酶标记山羊抗小鼠 IgG(H+L),北京中杉金桥生物技术有限公司;吐温 20,上海阿拉丁生化科技股份有限公司.
1.2 锚定蛋白的基因克隆
1.2.1cotC、cotG基因及treS基因的克隆
检索GenBank中已登录的cotC和cotG基因序列,通过软件CE Design V1.04设计多片段无缝克隆特异性引物,以本实验室构建质粒pET-15b-treS为模板扩增获得treS基因序列,以B. subitilisWB800n基因组 DNA为模板扩增cotC和cotG基因序列.将PCR扩增所得的cotC、cotG基因片段及其对应的treS基因片段,利用多片段无缝克隆技术分别连接获得融合基因cotC-treS及cotG-treS序列.所用引物见表 1,下划线为酶切位点.PCR 扩增条件为:95℃预变性3min,95℃变性15s,63℃退火15s,72℃延伸90s,30个循环;72℃延伸 5min,4℃保存.将 PCR产物通过琼脂糖凝胶电泳进行检验,并使用 SanPrep柱式DNA胶回收试剂盒进行胶回收,-20℃保存.
1.2.2 双cot-treS展示体系中的基因克隆
根据 1.2.1节所述融合基因 cotC-treS及 cotG-treS序列,用1.2.1节方法进行双cot-treS展示体系中的基因克隆.

表1 引物序列Tab. 1 Primer sequence
1.3 pDG1730-cotC/G-treS和pDG1730-cotC-treS-cotG-treS重组质粒的构建
根据 1.2.1节所述,获得 cotC、cotG基因及其对应的treS基因.根据整合型质粒pDG1730的HindⅢ和 BamHⅠ酶切位点,用这两个酶将 pDG1730质粒双酶切,并利用多片段无缝克隆技术将 cotC和 treS基因、cotG和 treS基因连接到 pDG1730质粒上,获得 pDG1730-cotC/G-treS重组质粒.再进一步利用多片段无缝克隆技术,将获得的 cotC-treS和 cotG-treS基因片段连接到经过 BamHⅠ和 HindⅢ双酶切的pDG1730质粒上,获得重组质粒pDG1730-cotC-treS-cotG-treS.构建流程如图1所示.
1.4 B. subitilis WB800n感受态的制备与电转化
将B. subitilis WB800n在LB平板中划线,长出单菌落后,挑取一个单菌落接种于 5mL LB液体培养基中,过夜培养.取 500µL过夜培养的菌液转接至 50mL GM 培养基中,使 A600=0.1~0.2,37℃、200r/min培养至 A600=1.0.将 GM 培养基及所有枪头、离心管置于冰上冷却10min.将GM培养基转移至 50mL离心管,4℃、5000r/min离心 8min.用20mL ETM 培养基重悬菌体,4℃、5000r/min离心8min,重复 3次.将洗涤后的菌体重悬于 500µL ETM 中,分装到 1.5mL离心管中,每管 60µL.在60µL感受态中加入6µL重组质粒,冰浴5min,加入到预冷的电转杯(2mm)中电击 1次(电压 2500V,时间 5.0ms).电转完毕后,加入 1mL 37℃预热的RM 培养基,转入 1.5mL离心管中,37℃、200r/min复苏 3h.将复苏后的菌液离心,除去多余上清液,留100µL上清液重悬菌体,涂布在含有壮观霉素(终质量浓度 100µg/mL)的 LB 平板上,37℃培养箱中倒置培养.

图1 重组质粒pDG1730-cotC-treS-cotG-treS的构建Fig. 1 Construction of recombinant pDG1730-cotC-treS-cotG-treS
1.5 芽胞表面展示TreS的分析
1.5.1 免疫印迹(Western blot)分析
将重组菌于含壮观霉素的 LB平板(终质量浓度100µg/mL)上划线活化,挑取单菌落至50mL含有壮观霉素的 LB液体培养基中过夜培养.按 1%的接种量接种于 TB培养基中,37℃、200r/min培养96h.7500r/min离心 10min,2mg/mL溶菌酶 37℃处理 30min后沉淀芽胞.取适量芽胞溶于 5mL的SDS-DTT溶液中,置于 37℃水浴中保温 2h.用50mmol/L Tris-HCl(pH 7.5)洗涤3次,最后用裂解液悬浮.将离心管置于冰上进行超声破碎(功率300W,工作 2s,间隔 4s,工作总时间为 15min).将破碎后的溶液在 4℃、8000r/min离心 20min,收集芽胞,用50mmol/L Tris-HCl(pH 7.5)重悬沉淀,收集上清液(上清液即为提取的含有 His tag的芽胞衣壳蛋白溶液),进行 SDS-PAGE电泳检测或将上清液浓缩后进行SDS-PAGE检测.
将提取的芽胞衣壳蛋白经 SDS-PAGE分离后,用电转移法将凝胶上的蛋白转移至 PVDF膜上,然后用 5%的脱脂奶粉室温下封闭 1h,将膜上封闭液洗净,于 4℃条件下一抗孵育过夜.回收一抗,用TBST缓冲液洗膜3次,每次5min.将封闭液稀释的二抗加到膜上,室温孵育 1h.回收二抗,用 TBST洗膜3次,每次10min.最后将膜放入显色液中显色后扫描.
1.5.2 免疫荧光分析
将重组菌转接至DSM培养基中37℃、200r/min培养 48h.取 48h培养物 2mL,12000r/min离心10min收集菌体,重悬于相同体积的 GTE buffer(50mmol/L 葡萄糖,10mmol/L EDTA,20mmol/L pH 7.5Tris-HCl,2mg/mL 溶菌酶)中,37℃处理 30min破坏营养细胞.12000r/min离心15min沉淀芽胞,用 pH 7.4的 PBS缓冲液(8g/L NaCl、0.24g/L KH2PO4、0.2g/L KCl、3.63g/L Na2HPO4·12H2O 溶于 800mL 蒸馏水中,HCl调 pH为 7.4,加水定容至 1L,121℃灭菌 25min,室温保存)洗涤 5次,用于免疫荧光分析.用 100µL抗体结合buffer(PBS中加入BSA和山羊血清使其体积分数都为3%)悬浮芽胞,在1.5mL EP管内操作,加入适当浓度的一抗,冰上放置 2h.用抗体结合缓冲液洗涤芽胞 4次,4000r/min离心 15min,完全除去上清液.用100µL抗体结合buffer悬浮芽胞,加入适当浓度FITC标记的二抗,冰上放置2h.用抗体结合缓冲液洗涤细胞 3次,5000r/min离心 10min,完全除去上清液.将 100µL悬液滴到载玻片上,盖上盖玻片,黑暗中待其吹干后立刻用共聚焦显微镜观察.
1.5.3 芽胞表面展示酶活力分析
将重组菌在含壮观霉素的LB平板上划线,将长出的单菌落接种于含壮观霉素的液体 LB中过夜培养后转接至TB培养基中37℃、200r/min培养96h,测芽胞干质量并进行芽胞计数.将芽胞离心,用 pH 8.0的PBS重悬芽胞,超声破碎(功率300W,间歇时间 5s,工作时间 5s,全程 15min).加入适量麦芽糖溶液使麦芽糖终体积分数为 30%,25℃条件下反应转化 1h.将转化反应后的样品煮沸 10min使酶失活,检测生成的海藻糖浓度,并计算海藻糖合酶的酶活力.酶活力单位(U)定义为在 25℃、pH 8.0、10mmol/L的磷酸缓冲体系中,1h转化生成 1µmol的海藻糖.
1.5.4 斑点印迹杂交(Dot blot)分析
利用 Dot blot分析方法对重组菌 B. subtilis WB800n/cotC-treS、B. subtilis WB800n/cotG-treS和 B.subtilis WB800n/cotC-treS-cotG-treS的芽胞所携带的TreS分子进行分析.用TBST溶液将TreS标准品溶液及提取的重组菌的芽胞衣壳蛋白溶液进行梯度稀释,成排点于 PVDF膜上并风干,记录好样品顺序,然后将风干好的 PVDF膜用封闭液室温振荡孵育1h,在室温下用一抗振荡孵育 1h,并用 TBST溶液洗涤 3次,每次 5min,接着再在室温下二抗振荡孵育1h,并用TBST溶液洗涤3次,每次10min,最后将膜放入显色液中显色后扫描,显色结果利用 Image J软件进行分析,输出结果的平均灰度值,转化为吸光度后进行阳性信号的定量化.在Excel软件中利用该公式计算各平均灰度值对应的吸光度,乘以所测区域的面积(Image J软件可同时给出),从而获得所测区域总吸光度,该值代表测量区域中物质的含量.
2 结果与分析
2.1 目的基因的克隆及重组质粒的构建
2.1.1 cotC、cotG基因及treS基因的克隆
以B. subtilis WB800n的基因组为模板,分别以cotC-F和cotC-R、cotG-F和cotG-R为引物进行PCR扩增,1%琼脂糖凝胶电泳检测,结果如图 2(a)所示.由图 2(a)可知,cotC基因在 500bp左右出现特异性条带,理论长度为408bp,cotG基因在750bp左右出现特异性条带,理论长度为 763bp,与理论长度相符,表明已成功获得 cotC和 cotG基因.以突变的pET15b-treS基因组为模板,分别以 cotC-treS-F-1/treS-R和cotG-treS-F-1/treS-R为引物进行PCR扩增,1%琼脂糖凝胶电泳检测,结果如图2(b)所示.由图2(b)可知,在2000bp左右出现特异性条带,与理论长度2085bp相符,表明已成功获得突变treS(C)、treS(G)基因片段.
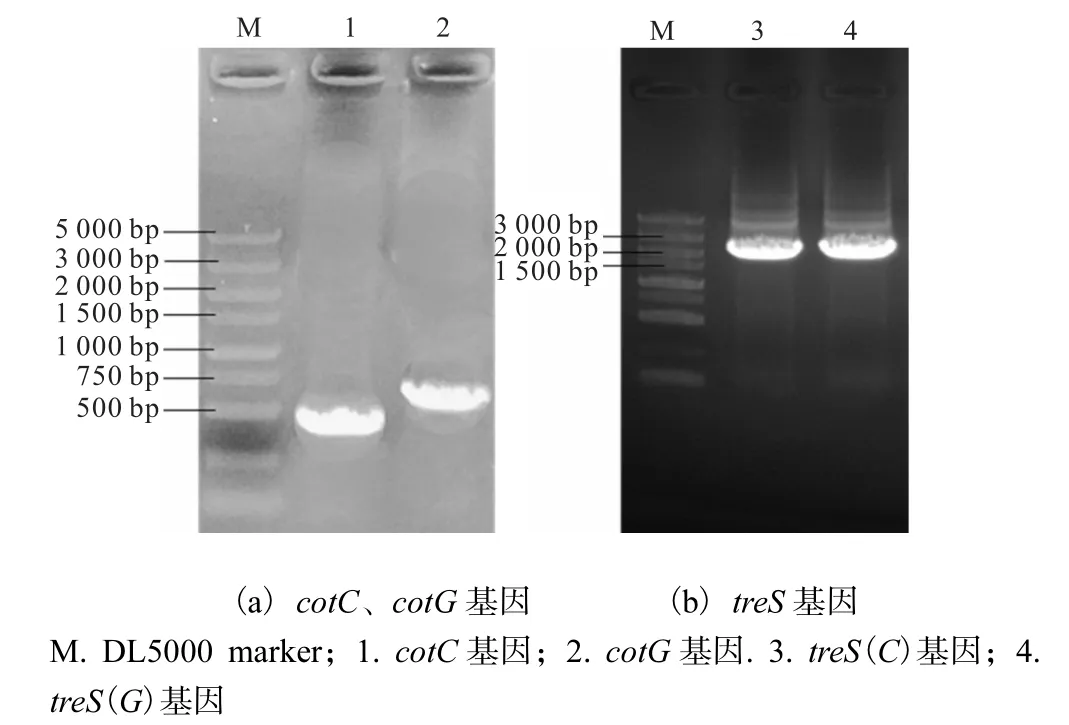
图2 cotC、cotG基因及treS基因的PCR扩增电泳图Fig. 2 PCR amplification electrophoresis of cotC,cotG and treS genes
2.1.2 重组质粒pDG1730-cotC/cotG-treS的构建
将PCR扩增所得的cotC、cotG基因及其对应的treS基因,利用多片段无缝克隆技术连接到pDG1730质粒中,并转化至E. coli DH5α感受态细胞中.利用特异性引物进行菌落 PCR验证,通过 1%琼脂糖凝胶电泳对 PCR产物进行检测.检测结果如图 3所示,表明cotC/cotG -treS基因已成功连接到pDG1730质粒.

图3 重组质粒pDG1730-cotC/cotG-treS的菌落PCRFig. 3 PCR of recombinant plasmid pDG1730-cotC/cotG-treS
2.1.3 双cot-treS体系中基因的克隆
以重组质粒 pDG1730-cotC-treS为模板,以cotC-treS-F-2和 cotC-treS-R-2为引物进行 PCR扩增,1%琼脂糖凝胶电泳检测,结果如图4所示.cotC-treS基因在2493bp出现特异性条带,与cotC-treS基因理论长度相符,说明已成功获得cotC-treS基因.
以重组质粒 pDG1730-cotG-treS为模板,以cotG-treS-F-2和 cotG-treS-R-2为引物进行 PCR扩增,1%琼脂糖凝胶电泳检测,结果如图4所示.cotG-treS基因在 2847bp出现特异性条带并高于 cotC-treS基因,与cotG-treS基因的理论长度相符,说明已成功获得cotG-treS基因.

图4 双 cot-treS体系中 cotC-treS和 cotG-treS基因的PCRFig. 4 PCR of cotC-treS and cotG-treS genes in double cot-treS system
2.1.4 重组质粒pDG1730-cotC-treS-cotG-treS的构建
将PCR获得的cotC-treS基因和cotG-treS基因,利用多片段无缝克隆技术连接到整合型质粒pDG1730上,并转化至E. coli DH5α中.分别利用特异性引物 cotC-F和 cotC-treS-R-2、cotG-F和 cotG-treS-R-2对同一菌株进行菌落 PCR验证,1%琼脂糖凝胶电泳进行检测,检测结果如图5所示.由图5可知,2—5号菌同时扩增出特异性条带 cotC-treS和cotG-treS,表明 cotC-treS基因和 cotG-treS基因已成功连接至pDG1730质粒中.

图5 重组质粒pDG1730-cotC-treS-cotG-treS菌落PCRFig. 5 PCR of recombinant plasmid pDG1730-cotC-treS-cotG-treS
2.2 重组菌的构建
2.2.1 重组菌的筛选
将重组质粒电转化至B. subtilis WB800n感受态中,涂布至含有壮观霉素的 LB平板上.利用特异性引物进行菌落 PCR验证,1%琼脂糖凝胶电泳检测.整合型质粒pDG1730电转至B. subtilis WB800n中,目的基因在两侧α-淀粉酶同源臂的作用下,以双交换整合到 WB800n的染色体中.将 B. subtilis WB800n和重组B. subtilis WB800n同时点种在含有淀粉的 LB平板中,37℃过夜培养,如图 6(a)所示.喷洒碘液后,B. subtilis WB800n原始菌有明显的淀粉水解圈,而重组菌无明显的淀粉水解圈,表明目的基因成功整合染色体淀粉酶基因位点,如图 6(b)所示.
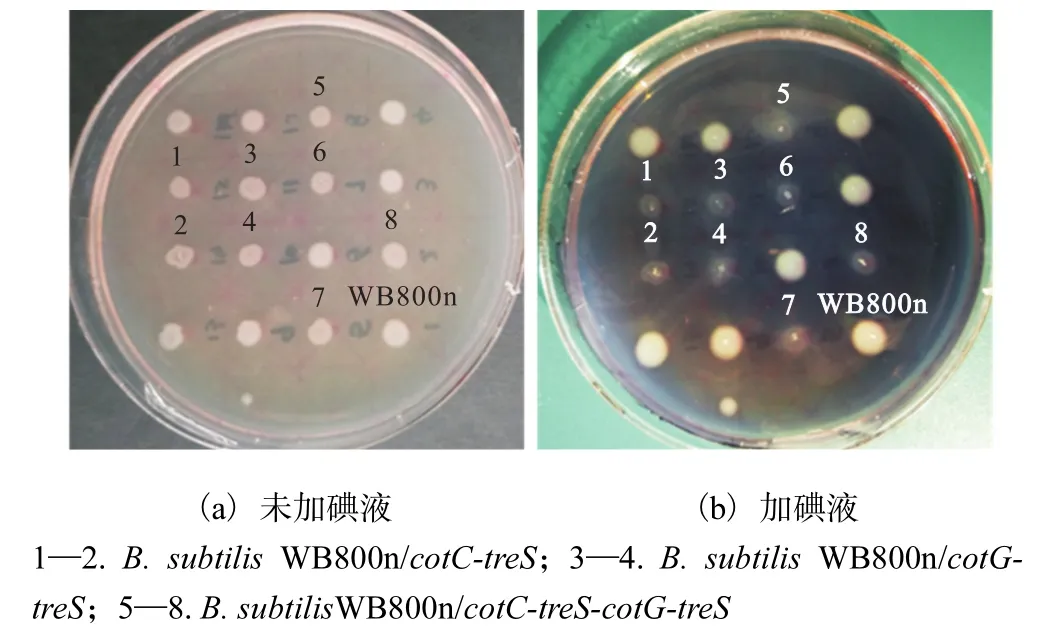
图6 重组菌的淀粉板验证Fig. 6 Starch plate validation of recombinant B. subtilis WB800n
2.2.2 芽胞表面展示TreS的Western blot分析
将重组菌 B. subtilis WB800n/cotC-treS、B. subtilis WB800n/cotG-treS、B. subtilis WB800n/cotC-treS-cotG-treS分别接种至 TB培养基中,培养 96h后收集芽胞并纯化.提取芽胞衣壳蛋白后,进行 Western blot分析,结果如图 7所示.由图 7可知,构建的重组菌芽胞分别成功表面展示 CotC-TreS、CotG-TreS及表面共展示CotC-TreS和CotG-TreS.图中1、2泳道条带分别对应的相对分子质量约为 9.95×104和8.44×104;3泳道无相应条带;4泳道条带为两条,分别对应 9.95×104和 8.44×104.这与衣壳蛋白 CotC和 TreS加和的理论相对分子质量、CotG与 TreS加和的理论相对分子质量相一致,表明芽胞表面分别成功展示了 CotC-TreS、CotG-TreS融合酶及共展示两者的融合酶.

图7 重组菌的芽胞衣壳蛋白Western blot分析Fig. 7 Analysis of the capsid protein Western blot of recombinant bacteria
2.2.3 芽胞表面展示TreS的免疫荧光分析
将重组菌接种至DSM培养基中,培养48h后收集芽胞.对芽胞进行处理后,荧光共聚焦显微镜进行观察,结果如图8所示.

图8 重组菌的免疫荧光分析Fig. 8 Immunofluorescence analysis of recombinant bacteria
出发菌株所产芽胞无荧光现象,重组菌所产芽胞均有荧光现象,且共展示重组菌芽胞荧光较单Cot展示重组菌芽胞荧光量明显增强,这说明重组菌成功将海藻糖合酶展示于芽胞表面,且共展示的海藻糖合酶量较单个展示的海藻糖合酶量有明显的优势.
2.2.4 芽胞表面展示TreS的Dot blot分析
取 107个重组菌芽胞,提取芽胞衣壳蛋白,进行Dot Blot分析,结果如图9所示.利用Image J软件对结果图像进行分析.通过对Dot blot结果图的分析可得 3种重组菌的单个重组芽胞中所展示的海藻糖合酶分子数.其中重组菌 B. subtilis WB800n/cotC-treS和B. subtilis WB800n/cotG-treS单个芽胞所展示的海藻糖合酶分子数分别为 4.318×109和 2.361×109,而重组菌B. subtilis WB800n/cotC-treS-cotG-treS单个芽胞所展示的分子数为 7.366×109.由此可知,双Cot共展示的蛋白分子数大于CotC和CotG展示分子数之和.

图9 表面展示TreS的Dot blot分析Fig. 9 Dot blot analysis of surface display of TreS
2.2.5 芽胞表面展示TreS的酶活力分析
芽胞表面展示 TreS的酶活力分析(以芽胞干质量计)结果如图 10所示.由图 10可知,重组菌 B.subtilis WB800n/cotG-treS的酶活力为484.51U/g,B.subtilis WB800n/cotC-treS的酶活力为 886.11U/g,B.subtilis WB800n/cotC-treS-cotG-treS的酶活力为1511.6U/g. 由此可知,B. subtilis WB800n/cotC-treS-cotG-treS的芽胞表面展示酶活力大于 B. subtilis WB800n/cotG-treS和B. subtilis WB800n/cotC-treS的芽胞表面展示酶活力之和.这一现象与Dot blot分析的 CotC与 CotG共展示在单个芽胞表面的 TreS酶分子数大于由单个Cot独立展示的TreS酶分子数之和相一致.在相同菌体细胞生成相同的芽胞数所呈现的单位菌体酶活力与这一现象一致,即单位酶分子的酶活力是相同的,而展现的酶活力增加主要是因为酶的表面展示的分子数增加造成的.
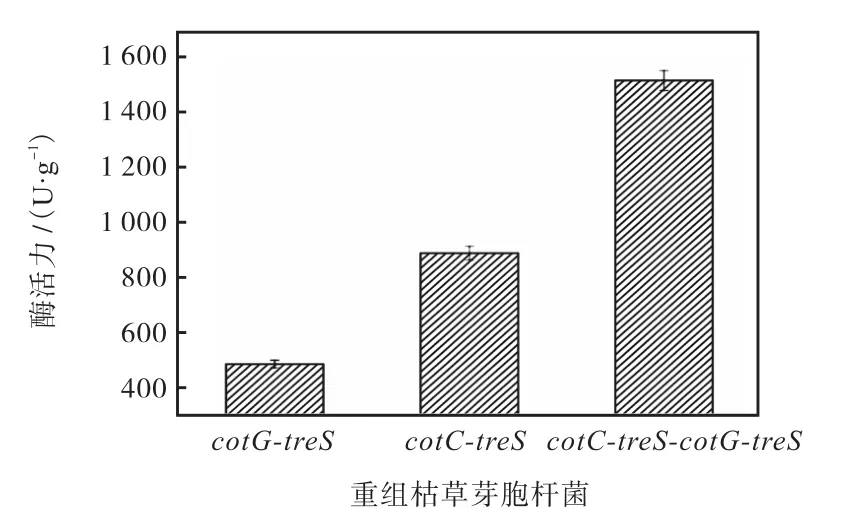
图10 芽胞表面展示TreS的酶活力Fig. 10 Enzyme activity of TreS on the surface of spare
3 结 论
枯草芽胞杆菌的芽胞表面有多种锚定蛋白,报道有多种锚定蛋白已成功用来展示外源蛋白.本实验通过CotC和CotG分别在芽胞表面成功展示了外源海藻糖合酶,并且通过 CotC和 CotG两种锚定蛋白在芽胞表面共展示了海藻糖合酶,经免疫荧光、Western blot、Dot blot和酶活力检测分析后,表明海藻糖合酶成功展示于芽胞表面,且双Cot共展示的效果较单Cot展示具有明显的增强效果.在TB培养基中,重组菌B. subtilis WB800n/cotC-treS-cotG-treS培养 96h后,芽胞表面展示的酶活力(以芽胞干质量计)达到 1511.6U/g,芽胞表面展示的酶分子数为7.366×109,大于CotC和CotG独立展示海藻糖合酶的酶活力(484.51U/g和 886.11U/g)及芽胞表面展示的酶分子数(4.318×109和 2.361×109)之和,该现象目前尚未有报道进行解释.这一结果给了我们一些启示:多个 Cot共展示外源蛋白时,可能存在一定的独立性,即不同的Cot蛋白在芽胞表面的展示分布具有自己独立的位点或在展示过程中具有区域性;当多Cot展示时,不同 Cot之间在转运和锚定过程中可能存在一种相互影响机制,这种机制或许具有增强作用,或许在其他 Cot和锚定蛋白间存在消减作用,有待进一步探索;不同Cot蛋白在芽胞表面展示外源蛋白的分子数之间的差异性,可能存在一个表面结合位点初始表达量或是转运过程中的多 Cot竞争性选择特性,有待进一步探索.本实验在给出上述启发的同时,也给外源酶的微固定化应用提供了一个思路,即通过芽胞这一微小的稳定载体,可以通过多种Cot展示的方法,提高芽胞表面展示外源酶、蛋白和疫苗的量,降低单位制备成本,提高效率,可以根据不同Cot间的芽胞展示分子数差异展示不同的外源酶,实现多酶的协同应用效果.
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
舰船科学技术(2022年11期)2022-07-15
中国生物防治学报(2022年3期)2022-07-09
湖北工业大学学报(2022年2期)2022-05-07
中国土壤与肥料(2021年5期)2021-12-02
福建农业科技(2021年4期)2021-08-02
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
食品工业科技(2014年21期)2014-03-11
