
水液相环境下羟自由基诱导的苯丙氨酸分子损伤机理
2019-12-19 08:43:48潘宇庄严姜春旭刘薛涛陶思宇佟华王佐成
浙江大学学报(理学版) 2019年6期
潘宇,庄严,姜春旭,刘薛涛,陶思宇,佟华*,王佐成*
(1.白城师范学院物理学院,吉林白城137000;2.白城师范学院计算机科学学院,吉林白城137000;3.白城师范学院传媒学院,吉林白城137000)
0 引言
苯丙氨酸(phenylalanine;Phe)是人体必需的氨基酸。按构型分为S-型苯丙氨酸(S-Phe)和R-型苯丙氨酸(R-Phe),按光学活性可分为左旋体(L-Phe)和右旋体(D-Phe)。L-Phe在生命体内具有活性,是构成蛋白质的重要成分,对控制肿瘤转移有一定的作用,还可用作人类食品添加剂[1]。D-Phe是重要的化工和生物制药原料[2]。生命体内氨基酸的消旋及其结构的改变可导致生物衰老[3-4]。
基于Phe的作用及其手性转变可导致生命体衰老,人们对其结构、光谱和旋光异构进行了广泛研究。文献[5]研究了气相Phe的构象,结果表明,其可以有多个稳定构象,氨基与羧基间具有分子内单氢键并且氨基与苯环间的π-氢键构象最稳定。文献[6]通过实验测量了L-Phe的红外和紫外光谱,解释了Phe具有分子内氢键。文献[7]实验测量了L-Phe的转动谱,进一步证实了L-Phe具有分子内氢键。文献[8-10]对其旋光异构进行了较系统的研究。研究表明,气相Phe在通常情况下具有稳定性;水分子簇的催化可使微量Phe实现手性对映体转变;水分子簇催化和水溶剂助催化的共同作用可使少量Phe分子实现手性对映体转变,解释了生命体内有DPhe存在。
生命体是富水环境,糟糕的情绪、剧烈的运动和紫外线的照射等会使体内产生大量的羟自由基。羟自由基的氧化能力极强,可以通过抽氢和加成等反应对氨基酸造成损伤[11]。文献[12]研究表明,羟自由基抽取α-氢可导致气相α-丙氨酸损伤。文献[13-14]的研究表明,羟自由基抽取α-氢可导致水液相环境下的α-丙氨酸损伤。文献[15]的研究表明,羟自由基抽取α-氢可导致赖氨酸损伤。文献[16-17]的研究表明,羟自由基抽取α-氢可导致缬氨酸损伤,单臂碳纳米管(SWCNT)对其损伤反应具有助催化作用,而水溶剂效应对其具有阻碍作用。Phe分子的R基含有苯环、亚甲基和次甲基,羟自由基诱导Phe损伤的研究具有一定的挑战性,目前尚未见相关研究的报道。本工作对研究其他含有苯环的有机分子(数量极其庞大)的损伤也具有积极的意义。基于此,对水液相环境下羟自由基(水分子簇)诱导的Phe损伤进行了研究,期望在理论上对羟自由基(水分子簇)致Phe分子损伤给予解释,同时告诉人们应养成良好的生活与工作习惯,尽量降低体内的羟自由基含量。
1 研究与计算方法
采用能较好地处理氢键等弱作用的色散校正密度泛函的WB97X-D[18]方法,结合处理溶剂效应的自洽反应场理论的SMD模型[19]方法,在WB97XD/SMD/6-311++G(d,p)水平优化反应过程的驻点构象,对过渡态[20]进行IRC(内禀反应坐标)[21]计算,验证其确实连接所期望的局域极小点,在同一水平下对体系进行自然键轨道(NBO)分析,获得其原子自然电荷(NPA)。为获得更精确的反应过程的势能面,采用微扰理论的MP2[22]方法,在MP2/SMD/6-311++g(3df,2pd)水平计算驻点的单点能,用零点振动能校正与单点能之和作为总能量。本工作选取S-型Phe分子作为目标反应物,研究羟自由基诱导其损伤。S-Phe分子与羟自由基水分子簇形成的复合物,标记为S-Phe·[·OH)·H2O],其他体系的标记相似。计算工作由Gaussian 09[23]程序在白城师范学院理论计算中心的服务器集群完成。
2 结果与讨论
水液相环境下S-Phe分子的最稳定构象[11]见图1。研究发现,羟自由基(水分子簇)抽取α-氢(α-碳3C上的氢)、β-氢(β-碳5C上的氢)、苯环-氢以及羟自由基与苯环加成均可致S-Phe分子损伤,下面分别进行讨论。
2.1 羟自由基(水分子簇)抽α-氢致S-Phe损伤
羟自由基(水分子簇)抽α-氢[NPA电荷计算表明,本文羟自由基(水分子簇)抽氢,均为抽“氢原子”,相似之处不再解释],可分为羟自由基水分子簇与α-氢和氨基作用、羟自由基水分子簇与α-氢和羰基作用以及羟自由基与α-氢作用形成反应复合物,而后羟自由基抽氢的3种情况。对应的3个抽氢反应通道,分别命名为a、b、c,反应历程见图2,反应势能面剖面见图3,下面分别进行讨论。

图2 羟自由基(水分子簇)抽α-H导致S-Phe损伤的反应历程(键长:nm)Fig.2 S-Phe damage mechanism induced by α-H abstraction of hydroxyl radicals(water clusters)(bond length:nm)
2.1.1 a通道
在a通道,损伤的反应历程见图2(a),反应势能面剖面见图3的a曲线。首先是反应物S-Phe的α-氢和氨基氢分别与羟自由基水分子簇的羟自由基和水分子作用,形成底物复合物a_S-Phe·[(·OH)·H2O](α-氢的序号由4变为23了)。而后底物复合物a_S-Phe·[(·OH)·H2O]经·OH抽α-氢 23H的过渡态a_TS·[(·OH)·H2O],异构成产物复合物 a_P*·(H2O)2。最后,产物复合物 a_P*·(H2O)2的a_P*和(H2O)2的分子间氢键解离,形成a_P*和二聚水(H2O)2。结构分析表明,a_P*·(H2O)2的1N—3C键长较短,小于a_S-Phe·[(·OH)·H2O]的1N—3C键长,1N、3C、5C、9O和21O形成了共轭的五中心大π键,水分子簇内以及水分子与氨基氢之间存在较强的氢键,水分子与α-碳以及水分子和羰基氧21O之间存在弱氢键,因此,a_P*·(H2O)2的构象稳定,处在很深的势阱中。a_P*是产物自由基,其α-碳上失去了氢原子,α-碳的手性已不存在,亦即a_P*失去了的手性,是损伤的Phe(后面相似之处不再解释)。
从a_S-Phe·[(·OH)·H2O]到a_TS·[(·OH)·H2O]过程,3C—23H键长从0.109 5 nm拉伸至0.123 0 nm断裂,反应活性中心骨架二面角1N—3C—4C—5C从125.29°变为129.32°,此过程化学键拉伸较小,活性中心骨架形变不明显,所需能量不高,3C—1N键长从0.146 0 nm收缩至0.143 7 nm,又会释放能量,过渡态的键角3C—23H—24O为176.67°,接近平角,3C—23H—24O具有较强的氢键,说明过渡态较稳定,因此 a_TS·[(·OH)·H2O]产生的内禀能垒非常低,只有6.7 kJ·mol-1。在此过程中,(·OH)·H2O中的H2O分子没有参与反应,其作用是稳定·OH的空间位置,使·OH处于抽取α-H原子的有利位置。从势能剖面可以看出,a_S-Phe·[(·OH)·H2O]不稳定,从反应物 S-Phe+[(·OH)·H2O]到反应复合物 a_S-Phe·[(·OH)·H2O]过程要吸热 61.7 kJ·mol-1。a_P*和(H2O)2之间的氢键解离能为81.6 kJ·mol-1,产物复合物 a_P*·(H2O)2可通过碰撞过程将其解离成a_P*和(H2O)2。
由于a_S-Phe·[(·OH)·H2O]不稳定,也可将该基元反应视为·OH直接抽氢机理,没有形成反应复合物的过程。从反应物到产物复合物是(·OH)·H2O的·OH直接抽氢原子23H形成H2O,亦即3C—23H键的断裂和24O—23H键的形成同时完成,反应能垒为68.4 kJ·mol-1。
2.1.2 b通道
在b通道,损伤历程见图2(b),反应势能面剖面见图3的b线。首先,羟自由基水分子簇与S-Phe的23H和9O作用,形成复合物b_S-Phe·[(·OH)·H2O]。而后b_S-Phe·[(·OH)·H2O]经·OH 抽取23H的过渡态 b_TS·[(·OH)·H2O],异构成产物复合物 b_P*·(H2O)2。最后b_P*和(H2O)2的分子间氢键解离,形成b_P*和二聚水(H2O)2。结构分析表明,b_P*·(H2O)2的1N、3C、5C、9O和21O形成了五中心共轭大π键,水分子间以及水分子与羰基氧间存在较强的氢键,水分子与α-碳之间存在弱氢键,其构象稳定。
从b_S-Phe·[(·OH)·H2O]到b_TS·[(·OH)·H2O]过程,3C—23H键长从0.109 3 nm拉伸至0.117 2 nm断裂,骨架二面角1N—3C—4C—5C从123.63°变为127.19°,化学键拉伸较小,活性中心骨架形变不明显,不需要太多的能量,过渡态的键角3C—23H—24O是172.64°,接近平角,3C—23H—24O具有较强的氢键,过渡态较稳定,因此b_TS·[(·OH)·H2O]产生的内禀能垒很低,只有29.5 kJ·mol-1。在此过程中,H2O分子也没有参与反应,其作用是稳定·OH的空间位置。从势能剖面中可以看出,从反应物 S-Phe+[(·OH)·H2O]到反应复合物 b_S-Phe·[(·OH)·H2O]的过程要吸热54.7 kJ·mol-1。b_P*和(H2O)2之间的氢键解离能是26.5 kJ·mol-1。同 2.1.1 节,b_S-Phe·[(·OH)·H2O]不稳定,该基元反应也可视为·OH直接抽氢,没有形成反应复合物的过程。·OH直接抽23H形成H2O,3C—23H键的断裂反应能垒为84.2 kJ·mol-1。
2.1.3 c通道
在c通道,损伤的反应历程见图2(c),反应势能面剖面见图3的c曲线。首先是羟自由基与23H作用,形成复合物 c_S-Phe·(·OH)。而后 c_S-Phe·(·OH)经·OH 抽取23H的过渡态 c_TS·(·OH),异构成产物复合物c_P*·H2O。结构分析表明,c_P*·H2O的1N、3C、5C、9O和21O形成了五中心大π键,水分子与羰基氧间存在较强的氢键,c_P*·H2O构象稳定。最后,c_P*和H2O的分子间氢键解离。

图3 羟自由基(水分子簇)抽α-H导致S-Phe损伤反应的势能面Fig.3 Potential energy surfaces of S-Phe damage induced by by α-H abstraction of hydroxyl radicals(water clusters)
从c_S-Phe·(·OH)到c_TS·(·OH)过程,3C—23H键长从0.109 4 nm拉伸至0.115 9 nm断裂,骨架二面角 1N—3C—4C—5C 从125.30°变为125.86°,基本没变。化学键拉伸较小,不需要较多的能量,3C—1N键长从0.146 1 nm收缩至0.144 0 nm,释放能量,过渡态的键角3C—23H—24O为177.85°,可视为平角,3C—23H—24O具有很强的氢键,过渡态很稳定,因此c_TS·(·OH)产生的内禀能垒很低,只有16.0 kJ·mol-1。从势能面剖面可以看出,从反应物S-Phe+(·OH)到反应复合物c_SPhe·(·OH)的过程要吸热68.9 kJ·mol-1。c_P*和H2O之间的氢键解离能为79.8 kJ·mol-1。
同 2.1.1 节,由于c_S-Phe·(·OH)不稳定,该基元反应也可视为·OH直接抽氢机理,·OH直接抽氢原子23H形成H2O,3C—23H的断裂和24O—23H键的形成同时完成,反应能垒为84.9 kJ·mol-1。
计算表明,a_P*、b_P*和c_P*P*的构象相同,亦即Phe分子在a、b和c通道的损伤产物相同。
从图3中可以看出,羟自由基(水分子簇)抽α-H致S-Phe损伤在a、b和c 3个反应通道的能垒分别为68.4,84.2和84.9 kJ·mol-1,通道 a能垒最低,产物复合物以及产物的能量均是a通道最低,因此,羟自由基水分子簇抽α-H致S-Phe损伤反应的a通道是优势反应通道。
2.2 羟自由基(水分子簇)抽β-氢致S-Phe损伤
羟自由基(水分子簇)抽取β-氢可分为羟自由基水分子簇与β-氢和氨基作用以及羟自由基与β-氢作用形成反应复合物,对应的抽氢反应分为2个通道,分别命名为d和e。研究表明,羟自由基(水分子簇)抽取的β-氢可以是7H或8H,反应机理基本相同,为节省篇幅,本工作只讨论羟自由基(水分子簇)抽取7H的情形,反应历程见图4,反应势能面剖面见图5,下面分别进行讨论:
2.2.1 d通道
在d通道损伤的反应历程见图4(a),反应势能面剖面见图5的d曲线。
首先羟自由基水分子簇与S-Phe的7H和2H作用,形成反应复合物 d_S-Phe·[(·OH)·H2O]。而后d_S-Phe·[(·OH)·H2O]经·OH 抽取7H的过渡态d_TS·[(·OH)·H2O],异构成产物复合物 d_P*·(H2O)2。d_P*是失去一个β-氢的产物自由基,SPhe失去7H后,5C及其附近的碳上的电荷分布与S-Phe明显不同,NPA电荷计算表明,它们的β-碳、α-碳和苯环上碳的电荷分布相差明显,其他骨架和非骨架原子上的电荷分布也有不同程度的改变,d_P*已不具有Phe的特性,亦即S-Phe损伤了,后面e通道的与此相似之处不再述说。结构分析表明,d_P*·(H2O)2的1N、3C、6C、10O和22O形成了五中心大π键,水分子间存在较强的氢键,水分子与氨基间存在中等偏强的氢键,水分子与苯环间存在中等偏弱的π氢键,其构象较稳定。但由于d_P*·(H2O)2的五中心大π键强度和分子间氢键强度不及a_P*·(H2O)2和b_P*·(H2O)2,所以,d_P*·(H2O)2的稳定性不及a_P*·(H2O)2和b_P*·(H2O)2,在势阱的位置也不是很深。
从d_S-Phe·[(·OH)·H2O]到d_TS·[(·OH)·H2O],5C—7H键长从0.109 3 nm拉伸至0.114 4 nm断裂,骨架二面角11C—9C—5C—3C从-92.53°变为-81.80°,化学键拉伸较小,骨架形变较小,不需要太多能量,过渡态的键角5C—7H—24O为179.18°,接近平角,过渡态较稳定,因此,d_TS·[(·OH)·H2O]产 生的内禀能垒很低,只有27.3 kJ·mol-1。此过程 (·OH)·H2O中的H2O分子没有参与反应,其作用是稳定·OH的空间位置。从势能剖面中可以看出,从反应物 S-Phe+[(·OH)·H2O]到反应复合物d_S-Phe·[(·OH)·H2O]过程要吸热 62.0 kJ·mol-1。d_P*和(H2O)2之间的氢键解离能为11.0 kJ·mol-1。

图4 羟自由基(水分子簇)抽β-氢导致S-Phe损伤的反应历程(键长:nm)Fig.4 S-Phe damage mechanism induced by β-H abstraction of hydroxyl radicals(water clusters)(bond length:nm)

图5 羟自由基(水分子簇)抽β-氢导致S-Phe损伤反应的势能面Fig.5 Potential energy surfaces of S-Phe damage induced by by β-H abstraction of hydroxyl radicals(water clusters)
同 2.1.1 节,d_S-Phe·[(·OH)·H2O]不稳定,该基元反应也可视为·OH直接抽氢,·OH直接抽β-氢原子7H形成H2O,5C—7H键的断裂和24O—7H键的形成同时完成,反应能垒为89.3 kJ·mol-1。
2.2.2 e通道
在e通道,损伤的反应历程见图4(b),反应势能面剖面见图5的e曲线。首先是羟自由基与7H作用,形成反应复合物 e_S-Phe·(·OH)。而后 e_SPhe· (·OH)经·OH抽 7H的过渡态 e_TS·(·OH),异构成产物复合物e_P*·H2O。结构分析表明,e_P*·H2O的1N、3C、6C、10O和22O形成了五中心大π键,水分子与氨基间存在中等强度的氢键,水分子与苯环之间存在中等偏弱的π氢键,其构象较稳定。但由于e_P*··H2O的五中心大π键及氢键的强度不及c_P*·H2O,所以,e_P*·H2O的稳定性不及c_P*·H2O。从e_S-Phe·(·OH)到e_TS·(·OH)过程,键长5C—7H从0.109 4 nm拉伸至0.115 5 nm断裂,骨架二面角11C—9C—5C—3C从-93.31°变为-81.60°,化学键拉伸较小、骨架形变较小,不需要太多能量,过渡态键角5C—7H—24O为171.21°,接近平角,过渡态较稳定,因此e_TS·(·OH)产生的内禀能垒很低,只有21.9 kJ·mol-1。从势能剖面中可以看出,从反应物S-Phe+(·OH)到反应复合物e_S-Phe·(·OH)的过程要吸热 69.1 kJ·mol-1。e_P*和·H2O之间的氢键解离能是15.5 kJ·mol-1。
同 2.1.1 节,e_S-Phe·(·OH)不稳定,该基元反应也可视为·OH直接抽氢,·OH直接抽β-氢7H形成H2O,5C—7H键的断裂和24O—7H键的形成同时完成,反应能垒为91.0 kJ·mol-1。
从图5中可以看出,羟自由基水分子簇抽β-氢和羟自由基抽β-氢致S-Phe反应能垒为89.3~91.0 kJ·mol-1,损伤反应在竞争中不分伯仲,在误差允许范围内可认为相等。但考虑到水液相环境下羟自由基水分子簇的分布要多于羟自由基,羟自由基水分子簇抽β-氢致S-Phe损伤反应在竞争中应占主导地位。
与2.1节的羟自由基(水分子簇)抽α-氢致SPhe损伤相比较,本节的羟自由基(水分子簇)抽β-氢致S-Phe损伤处于劣势。主要原因是α-C-H键长(0.109 3 nm)大于β-C-7H键长(0.109 2 nm),NPA电荷计算表明,α-C正电荷量(-0.135 e)小于β-7H(-0.410 e),α-C-H键比β-C-7H的库伦作用弱,α-C-H键比β-C-7H键弱,容易断裂;NPA电荷计算表明,α-H正电荷量(0.253 e)大于β-7H正电荷量(0.231 e),羟自由基的氧原子对α-H的库仑引力大于对β-7H的库仑引力。
2.3 羟自由基抽苯环-氢致S-Phe损伤
羟自由基抽苯环-氢致S-Phe损伤命名为f通道。研究发现,羟自由基可以抽取苯环上的17H、19H和20H致S-Phe损伤(由于受氨基和羧基的影响,羟自由基抽取苯环上的13H和16H反应过程的驻点没找到),分别命名为f1、f2和f3通道,各通道的反应历程见图6,反应势能面剖面见图7。
对于f1通道,首先是羟自由基与苯环-氢17H作用,形成反应复合物f1_S-Phe·(·OH)。而后f1_S-Phe·(·OH)经·OH 抽取氢原子 17H的过渡态f1_TS·(·OH),异构成产物自由基复合物f1_P*·H2O。结构分析表明,f1_P*·H2O的1N、3C、6C、10O和22O没形成大π键,f_1P*和H2O之间的作用比较弱,失去氢原子的苯环碳具有比较强的获取氢原子的能力,f1_P*·H2O和f_P*的构象都不稳定。
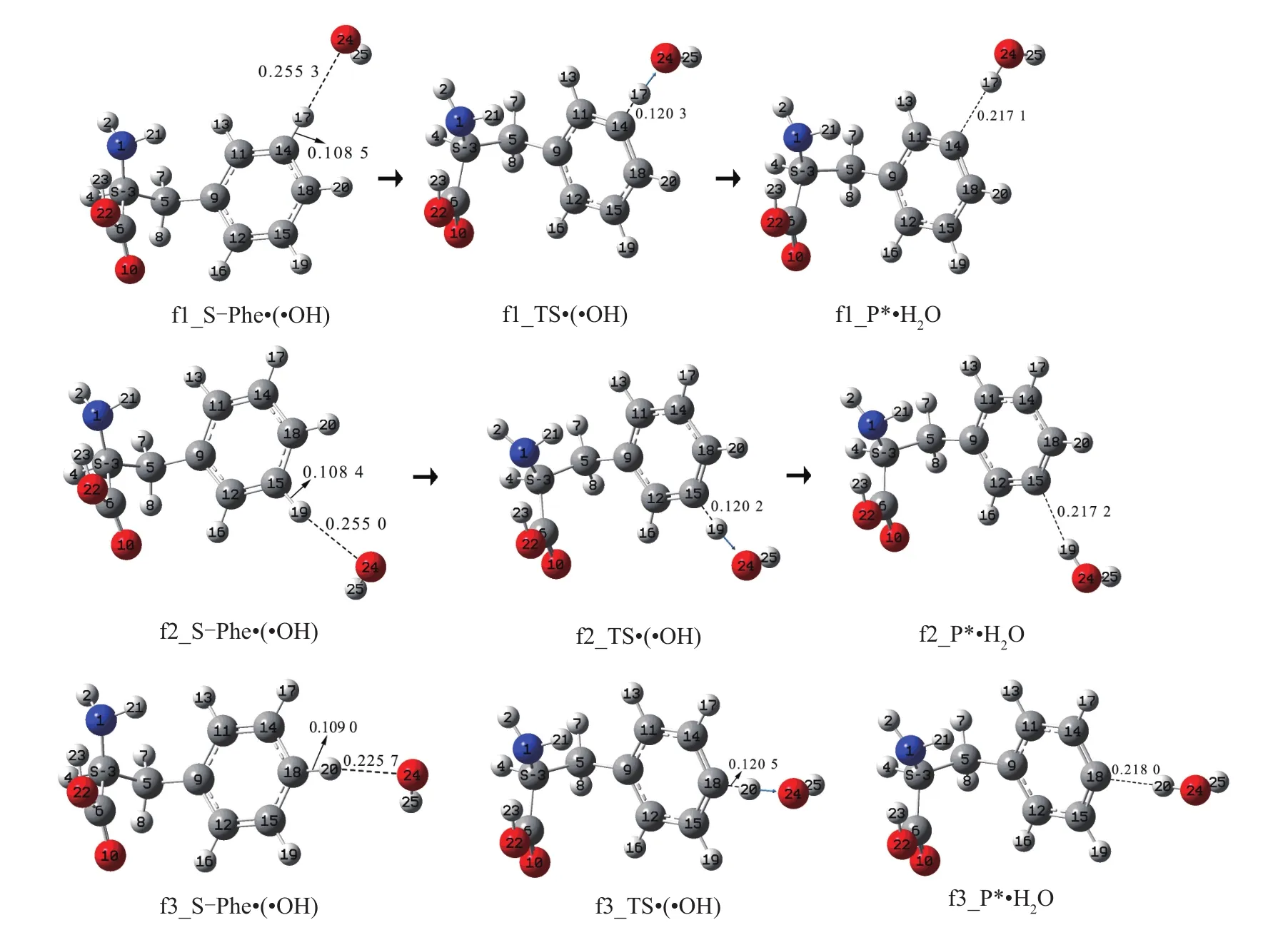
图6 羟自由基抽苯环-氢导致S-Phe损伤的反应历程(键长:nm)Fig.6 S-Phe damage mechanism induced by benzene ring-H abstraction of hydroxyl radicals(water clusters)(bond length:nm)
从f1_S-Phe·(·OH)到f1_TS·(·OH)过程,14C—17H键长从0.108 5 nm拉伸至0.120 3 nm断裂,化学键拉伸较小,所需能量较少,过渡态的键角14C—17H—24O为169.96°,接近平角,14C—17H—24O的氢键较强,过渡态较稳定,f1_TS·(·OH)产生的内禀能垒很低,只有7.0 kJ·mol-1。从势能面上可以看出,从反应物S-Phe+(·OH)到反应复合物f1_S-Phf·(·OH)吸热 104.6 kJ·mol-1,f1_P*和H2O 之间的氢键解离能为2.4 kJ·mol-1,f1_P*·H2O的分子间氢键作用极弱,f1_P*·H2O很容易通过碰撞解离成f1_P*和H2O。
同 2.1.1节,f1_S-Phf·(·OH)不稳定,该基元反应也可视为·OH直接抽氢,·OH直接抽取苯环-氢的原子17H形成H2O,14C—17H键的断裂和24O—17H键的形成同时完成,反应能垒为111.6 kJ·mol-1。
f3通道的机理类似于f1通道,从反应物SPhe+(·OH)到反应复合物 f3_S-Phe·(·OH)过程要吸热 107.9 kJ·mol-1,过渡态较 f3_TS·(·OH)产生的内禀能垒为4.7 kJ·mol-1,产物自由基f3_P*和H2O之间的解离能为0.7 kJ·mol-1,说明 f1_P*·H2O的分子间存在的是范德华作用,f1_P*·H2O很容易被解离为f1_P*和H2O。
f2通道不同于f1和f3通道,反应复合物f2_S-Phf·(·OH)极不稳定,其相对于反应物 S-Phe+(·OH)的能量为120.2 kJ·mol-1,比过渡态 f2_TS·(·OH)的能量还高,该通道的损伤反应在通常情况下很难实现,产物复合物f1_P*·H2O也极易解离。
从图7中可以看出,f2通道明显处于劣势,羟自由基抽取苯环-氢致S-Phe损伤反应的能垒为120.2 kJ·mol-1,如此高的能垒很难越过。在f1和f3通道,羟自由基抽取苯环-氢致S-Phe损伤反应的能垒分别为111.6和112.6 kJ·mol-1,与2.1和2.2节中的羟自由基(水分子簇)抽α-氢和β-氢致S-Phe损伤相比均处于劣势。主要原因有2个:(1)苯环的C—H键长短(苯环的C—H键长约为0.108 5 nm,α-C—H键长为0.109 3 nm,β-C—7H键长为0.109 2 nm),苯环的C—H键较强,不容易断裂;(2)苯环-H的正电荷量最小(NPA电荷计算表明,苯环-H正电荷量约为0.225 e,α-H正电荷量为0.253 e,β-7H正电荷量为0.231 e),羟自由基的氧原子对苯环-H的库仑引力最弱。从图7中还可看出,Phe在f1,f2和f3通道的损伤可自行修复。
2.4 羟自由基加成到苯环致S-Phe损伤
羟自由基与苯环加成可分为羟自由基在Phe分子内侧和外侧加成到苯环碳上2种情况,对应的反应通道分别命为g和h。
2.4.1 羟自由基在S-Phe内侧加成到苯环

图7 羟自由基抽苯环-氢导致S-Phe损伤反应的势能面Fig.7 Potential energy surfaces of S-Phe damage induced by benzene ring-H abstraction of hydroxyl radicals(water clusters)

图8 羟自由基加成到苯环C导致S-Phe损伤的反应历程(键长:nm)Fig.8 S-Phe damage mechanism induced by the addition of hydroxyl radicals to benzene ring C(bond length:nm)
研究表明,羟自由基在内侧加成到14C和15C位点的情况基本相同,而在9C、11C和12C的加成没有找到理想的驻点,本节只讨论羟自由基加成到14C和18C位点的情况,分别命名为g1和g2通道,反应历程见图8(a),反应势能面见图9(a),下面分别进行讨论。
对于g1通道:首先羟自由基与14C作用,形成复合物 g1_S-Phe·(·OH)。而后 g1_S-Phe·(·OH)经·OH加成到14C的过渡态g1_TS·(·OH),异构成产物g1_P*。g1_P*与S-Phe的结构明显不同,NPA电荷计算表明,它们在苯环上的电荷分布相差明显,g1_P*是损伤了的S-Phe。后面相似之处不再赘述。

图9 羟自由基加成到苯环导致S-Phe损伤反应的势能面Fig.9 Potential energy surfaces of S-Phe damage induced by the addition of hydroxyl radicals to benzene ring C
从g1_S-Phe·(·OH)到g1_TS·(·OH)过程,14C—24O键长从0.262 8 nm压缩至0.210 3 nm,需外界提供能量克服库仑斥力做功,骨架二面角18C—14C—15C—12C从179.15°变为176.21°,碳氧键压缩不是很大,需要的能量不很高,因此,g1_TS·(·OH)产生了33.2 kJ·mol-1的内禀能垒。从势能剖面中可以看出,从反应物S-Phe+(·OH)到复合物g1_S-Phe·(·OH)过程要吸热 73.5 kJ·mol-1。g1_SPhe·(·OH)不稳定,该基元反应也可视为不经过反应复合物,·OH直接经过渡态加成到14C,反应能垒为106.7 kJ·mol-1。
对于g2通道,首先羟自由基与18C作用,形成复合物 g2_S-Phe·(·OH)。而后 g2_S-Phe·(·OH)经·OH加成到18C的过渡态g2_TS·(·OH),异构成产物 g2_P*。g2_TS·(·OH)产生了43.2 kJ·mol-1的内禀能垒。g2_TS·(·OH)产生的内禀能垒大于g1_TS·(·OH)产生的,原因是从g2_S-Phe·(·OH)到g2_TS·(·OH)过程,18C—24O键长从0.278 6 nm压缩至 0.210 3 nm,与从g1_S-Phe·(·OH)到g1_TS·(·OH)过程14C—24O键长的压缩情况相比较,末状态键长 18C—24O 相同,初状态 g2_S-Phe·(·OH)的18C—24O 距离远,从g2_S-Phe·(·OH)到g2_TS·(·OH)过程需提供抵抗静电力做功的能量要多。
类似于g1通道,该基元反应也可视为不经过反应复合物,·OH直接经过渡态加成到18C,反应能垒为110.2 kJ·mol-1。
2.4.2 羟自由基在S-Phe外侧加成到苯环
研究表明,羟自由基在外侧加成到11C、12C、14C和15C的情况基本相同,在9C的加成没有找到理想的驻点,在此只讨论羟自由基加成到14C和18C的情况,分别命名为h1和h2通道,反应历程见图8(b),反应势能面见图9(b),反应机理与在g1和g2通道的相似,只做一般讨论。
对于h1通道:首先是羟自由基与14C作用,形成反应复合物 h1_S-Phe·(·OH)。而后 h1_S-Phe·(·OH)经过渡态 h1_TS·(·OH),异构成产物 h1_P*。从h1_S-Phe·(·OH)到h1_TS·(·OH)过程,14C—24O键长从0.257 6 nm压缩至0.209 8 nm,h1_TS·(·OH)产生的内禀能垒为31.7 kJ·mol-1。从势能剖面中可以看出,从反应物S-Phe+(·OH)到反应复合物 h1_S-Phe·(·OH)的过程要吸热76.8 kJ·mol-1。该基元反应也可视为不经过反应复合物,·OH直接经过渡态加成到14C,反应能垒为108.5 kJ·mol-1。
对于h2通道,先是羟自由基与18C作用,形成复合物 h2_S-Phe·(·OH)。而后 h2_S-Phe·(·OH)经过渡态 h2_TS·(·OH),异构成产物 h2_P*。h2_TS·(·OH)产生的内禀能垒为25.3 kJ·mol-1。h2_TS·(·OH)较 h1_TS·(·OH)产生的内禀能垒小。原因是从h2_S-Phe·(·OH)到h2_TS·(·OH)过程中,18C—24O的压缩距离小,并且h2_S-Phe·(·OH)的键长18C—24O 小于h1_S-Phe·(·OH)的键 长 18C—24O,h2_TS·(·OH)的键长 18C—24O 大于h1_TS·(·OH)的键长18C—24O。从h2_S-Phe·(·OH)到h2_TS·(·OH)需提供抵抗静电力做功的能量少。该基元反应也可视为不经过反应复合物,·OH直接经过渡态加成到18C,反应能垒为106.5 kJ·mol-1。从图9中可以看出,在误差允许的范围内,羟自由基在苯丙氨酸的内侧和外侧加成到14C和18C致SPhe反应损伤的能垒基本相同。从图9中还可以看出,这几个反应的产物基本与反应物处于势能面的同一高度。因此,羟自由基在苯丙氨酸的内侧和外侧加成到14C和18C致S-Phe损伤的反应在竞争中不分伯仲。
3 结 论
3.1 羟自由基诱导的S-Phe分子损伤反应有羟自由基(水分子簇)抽取α-氢、β-氢、苯环-氢以及羟自由基与苯环加成4种途径。这4种途径又分别有a、b和c通道,d和e通道,f1、f2和f3通道;g1、g2、h1和h2通道。
3.2 在a、b和c通道的竞争中,a通道具有优势,反应的能垒为68.4 kJ·mol-1,放热 98.0 kJ·mol-1。d和e通道的竞争不分伯仲,反应的能垒在89.3~91.0 kJ·mol-1,放 热在 54.9~57.3 kJ·mol-1。f1、f2和f3 反应通道的能垒在111.6 kJ·mol-1左右,是吸热反应。g1、g2、h1和h2通道的竞争也不分伯仲,反应能垒在106.5~110.2 kJ·mol-1,吸放热情况不明显。在所有的反应通道竞争中,a通道最具优势。
结果表明,羟自由基(水分子簇)抽取α-氢是Phe分子损伤的主要途径。
猜你喜欢
吉林大学学报(理学版)(2024年5期)2024-01-01 00:00:00
燃料化学学报(2023年3期)2023-03-11 03:34:40
中学化学(2022年5期)2022-06-17 16:51:48
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
高中数理化(2020年1期)2020-02-29 02:21:18
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
