
VPS35对多巴胺受体D1降解的调控机制研究
2019-11-09 02:19肖乃安马琪林詹奕红
中风与神经疾病杂志 2019年10期
王 琛,肖乃安,马琪林,詹奕红
帕金森病(Parkinson’s disease,PD)是一种常见的年龄相关的神经变性疾病,病情常呈慢性进行性加重,中枢神经系统中黑质纹状体等部位的多巴胺能神经元进行性退变、死亡,多巴胺合成或传导能力减弱,导致锥体外系主导的运动调节功能受损是其主要病理变化。神经递质多巴胺(dopamine)通过神经末梢上的受体结合,激活下游信号通路,参与多种神经系统的功能。多巴胺受体(Dopamine receptor)为7个跨膜区域组成的G蛋白偶联 (G protein-coupled)受体家族。目前根据对腺苷酸环化酶(adenylyl cyclase)的激活或者抑制作用已经鉴定出5种多巴胺受体亚型,分为D1类和D2类[1]。前者包括D1和D5受体;后者包括D2、D3、D4受体。囊泡分拣蛋白35(vacuolar protein sorting-35,VPS35)是Retromer复合体(VPS26、VPS29、VPS35和SNXs)的重要原件,主要负责分拣膜蛋白并介导其在胞内体至细胞膜及高尔基体之间的转运。有研究表明VPS35在帕金森患者的黑质等部位含量下降,且VPS35的D620N 突变与帕金森病发病相关。目前对VPS35缺陷(突变)参与帕金森病发病机制的研究主要集中在线粒体功能障碍,α突触核蛋白(α-synuclein)堆积以及神经元发育重塑缺陷等方面[2]。
VPS35能够调控CI-M6PR配体和cathepsin D的内吞再循环,促使α-synuclein通过溶酶体途径降解,进而减少其异常聚集[3]。除了经典的自噬溶酶体途径,VPS35还参与蛋白的泛素化降解(Ubiquitin proteasome),如VPS35能与Hrs相互作用[4],识别被泛素化修饰的受体,并分拣转运至溶酶体而降解[5]。在前期的实验中,我们发现过表达VPS35能够促进内吞的DRD1蛋白水平迅速下降,一方面,这可能是由于过表达VPS35促进了内吞的DRD1再循环上膜[6]。考虑到内吞蛋白还可能被转运到溶酶体降解,我们进一步研究VPS35是否影响DRD1的降解过程。依据CHX可以有效抑制生物体内蛋白的合成原理,我们用放线菌酮(cycloheximide,CHX)处理细胞,来研究VPS35对DRD1的降解可能存在的影响。
1 实验和材料
1.1 细胞株及神经元 HEK 293T(human embryonic kidney 293T,人胚肾细胞):厦门大学神经科学研究所提供。
1.2 菌种 E.coli DH5α,厦门大学神经科学研究所提供。
1.3 培养基 (1)细胞培养基 HEK293T培养基:DMEM、10%FBS、1%双抗;(2)细菌培养基 大肠杆菌培养基:NaCl(10 g)、east Extract(5 g)、Tryptone(10 g)、琼脂糖粉(15 g)(固体时需加),加900 ml的单蒸水溶解,5 mol NaOH调pH值至7.0后定容至1 L,高压蒸汽灭菌120 ℃ 20 min,4 ℃保存。
1.4 质粒 pCMV-myc(哺乳动物表达载体,C端带有myc标签)、pCMV-HA(哺乳动物表达载体,C端带有HA标签)、DRD1-HA、shVPS35质粒均由厦门大学神经科学研究所构建并保存。VPS35-myc质粒由新加坡分子与细胞生物学研究提供。
1.5 抗体、试剂及化学药品
1.5.1 抗体 HA-tag (26D11)抗体:Abmart公司;VPS35 抗体:Millipore公司;DRD1、Normal mouse IgG及Myc(9E10)抗体:购自Santa Cruz公司;GAPDH抗体:Cell Signaling Technology公司。
1.5.2 主要试剂及化学药品 DMEM(Dulbecco’s Modified Eagle’s Medium):Thermo scientific,Hyclone公司;0.25% Trypsin-EDTA(1×),(100X) (Penicillin-Streptomycin Solution)双抗:Invitrogen公司;Neurobasal、FBS(Fetal Bovine Serum)、HBSS及Opti-MEM®I:Gibco公司。限制性内切酶、T4 DNA连接酶、DNA Ladder Marker和Premix PrimerSTAR HS:TaKaRa公司;Rever Tra Ace qPCR RT Kit:TOYOBO东洋纺公司;Protein Ladder:Piercenet公司;放线菌酮(Cycloheximide,CHX):Sigma-Aldrich公司;MG-132:selleck公司;0.22、0.45 μmol PVDF WB膜:Millipore公司;质粒大提试剂盒:omegascientific公司;医用X光感蓝胶片:白云三和公司;氯化铵、谷胱甘肽、N-N’亚甲基双丙烯酰胺、过硫酸铵、丙烯酰胺、SDS等分析纯均国产。
2 实验方法
2.1 实验所需质粒构建 (1)根据Pubmed基因库人源DRD1基因(Gene ID:1812)序列,用Primer Primer 5.0软件设计出pcmv-DRD1-HA正向引物:5’-CGGAATTCATGAGGACTCTGAACACCTCTG-3’(限制性内切酶EcoRI )以及反向引物5’-CCTCGAGTCAGGTTGGGTGCTGACCGTTTTGT-3’(XhoI);由广州英潍捷基生物公司合成。(2)采用PCR技术从人的胎脑cDNA(互补DNA)文库中克隆出DRD1的 cDNA,将克隆出的cDNA和pcmv-HA载体用限制性内切酶进行双向酶切;(3)用T4 DNA 连接酶连接酶切回收的DRD1 cDNA和线性化pcmv-HA载体,连接产物于E.coli DH5α感受态大肠杆菌细胞中转化,后涂于含氨苄西林抗生素的固体LB培养皿平板上,37 ℃正置30 min后倒置培养12 h~16 h;选取合适的单克隆菌落挑取接种于含氨苄西林的液体LB中,于37 ℃摇床上震荡培养12 h~16 h;取适量(1~2 μl)菌液进行PCR鉴定,将克隆呈阳性的菌液送至英潍捷基公司双向测通测序,结果正确的克隆阳性菌液于4 ℃储存备大提。(4)根据文献报道[27]已知人源VPS35:shRNA1:5’-TCAGAGGATGTTGTATCTTTACAAGTCTC-3’,shRNA2:5’-GCTTCACACTGCCACCTTTGGTATTTGCA-3’,干扰序列的靶基因mRNA水平干扰效果(见图1)。
2.2 DRD1降解实验 于HEK 293T细胞中瞬时转染DRD1-HA质粒,4 h~6 h后等量传代至100 mm培养盘中。24 h后瞬时转染等量的对照载体pcmv-myc和VPS35-myc质粒,或等量的对照shRNA和VPS35-sh1,4 h~6h后等量传代至6-well培养盘中继续培养24 h后加500 μmol CHX,根据实验设计分别处理0 h、1 h、2 h、4 h、6 h和8 h(用DMSO作为0 h对照),TNEN裂解细胞并Western blotting检测目的蛋白的表达量,分别用myc和VPS35抗体检测VPS35,HA抗体检测外源的DRD1-HA,以GAPDH为内参,胶图用Image J软件灰度定量分析,GraphPad Prism 5分析数据并作图,*P<0.05,**P<0.01。于HEK 293T细胞中瞬时转染等量的对照载体pcmv-myc和VPS35-myc质粒,4 h~6 h后等量传代至6-well培养盘中。18 h~24 h后瞬用500 μmol CHX、及20 μmol MG132或者30 mmol NH4CL处理6 h(用DMSO作为0 h阴性对照),TNEN裂解细胞并Western blotting检测目的蛋白的表达量,分别用myc标签抗体检测VPS35,HA抗体检测外源的DRD1-HA,以GAPDH为内参,胶图用Image J软件灰度定量分析,GraphPad Prism 5分析数据并作图,*P< 0.05,**P<0.01,***P<0.001,n.s.:not significant。(每组用DMSO处理的DRD1蛋白量定为1)。
2.3 统计学分析 需要定量数据均为3个或3个以上的独立实验结果取平均值,用Image J软件进行灰度定量分析;用Graphpad prism 5统计软件作图统计分析,显著性差异由unpairedttest或paired检验确定,用“*”符号表示:*P<0.05,**P<0.01,***P<0.001。
3 结 果
我们用CHX处理细胞,来研究VPS35对DRD1的降解可能存在的影响。首先在稳定表达外源性带HA标签的人源DRD1蛋白的HEK293T细胞中瞬时转染对照载体pcmv-myc和VPS35-myc质粒(见图2A),4 h~6h后等量传代至6-well培养盘中继续培养24 h后加500 μmol CHX,根据实验设计分别处理0 h、1 h、2 h、4 h、6 h和8 h(用DMSO作为0 h对照),TNEN裂解细胞并Western blotting检测目的蛋白的表达量。结果显示在过表达VPS35之后,DRD1蛋白的降解速率比对照组加快了,说明VPS35促进了DRD1的降解。相反的,在下调VPS35水平后(见图2B),DRD1蛋白的降解速率比对照组减慢了。这些结果表明,VPS35能够调控DRD1的降解。
为了更深入地了解VPS35对DRD1降解的调控方式,我们采用20 μmol MG132和30 mmol NH4CL处理细胞作为阳性对照,DMSO作为0 h阴性对照(见图2C)的降解实验来分析VPS35调控DRD1降解的途径。用TNEN裂解细胞并Western blotting检测目的蛋白的表达量。结果显示,在转Vector细胞中,NH4Cl和MG132处理组的DRD1蛋白量比对照组都有所升高,但NH4Cl组升高更明显,提示DRD1在正常情况下主要通过溶酶体途径降解,这与七次跨膜蛋白的降解特点相吻合。而在转VPS35细胞中,NH4Cl和MG132处理组的DRD1蛋白量比对照组的升高趋势较Vector组都有所增加,提示VPS35过表达时,能增加DRD1的溶酶体和泛素蛋白酶体降解,但增加溶酶体降解的效果更加显著,因此抑制溶酶体才能更多地恢复其水平。
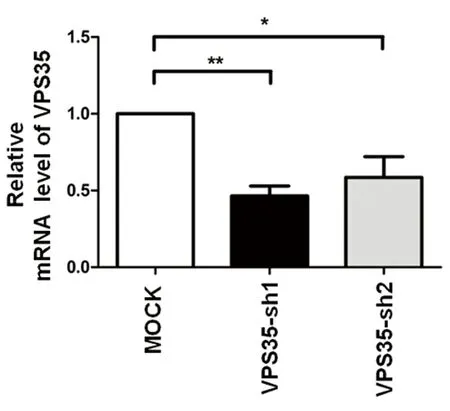
注:HEK293T细胞中转染 hVPS35,shRNA及对照质粒。 72 h后收集细胞提RNA并反转录, β-actin作为内参,Real-time PCR检测hVPS35表达量*P<0.05,**P<0.01
图1 靶基因mRNA水平干扰效果
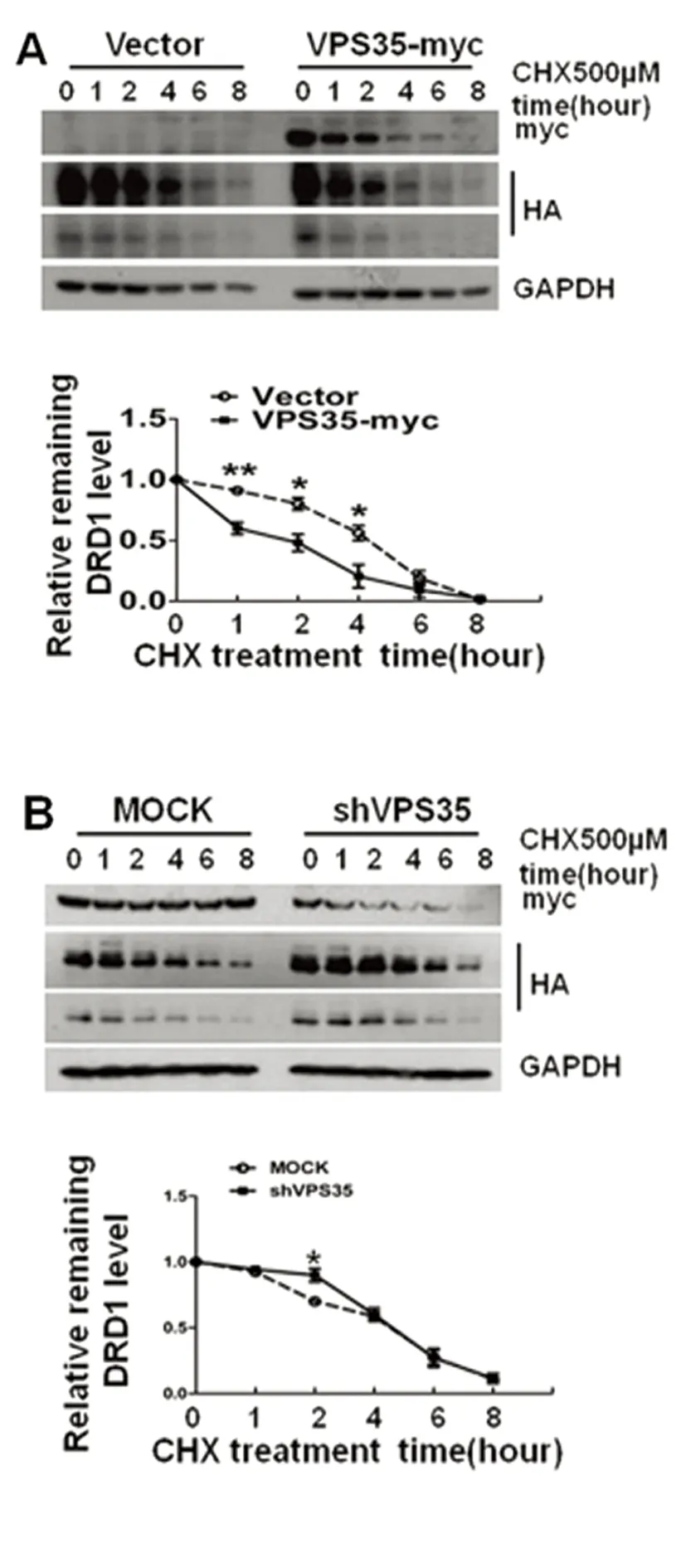

注:在过表达VPS35之后(A),DRD1蛋白的降解速率与对照组比较;在下调VPS35水平后(B),DRD1蛋白的降解速率与对照组比较;过表达VPS35、CHX及20 μmol MG132或者30 mmol NH4CL处理细胞(用DMSO作为0 h阴性对照)(C),DRD1蛋白的降解程度之间比较,以GAPDH为内参,胶图用Image J软件灰度定量分析,GraphPad Prism 5分析数据并作图*P<0.05,**P<0.01,***P<0.001,n.s.:not significant(每组用DMSO处理的DRD1蛋白量定为1)
图2 VPS35影响DRD1的降解
4 讨 论
帕金森病是一种病情呈进行性加重神经变性疾病,已成为威胁中老年人健康的“杀手”之一,PD多发于60岁以上的老年人群中,且低龄化趋势愈发加重。由于PD是中枢神经系统退行性疾病,黑质纹状体等部位的多巴胺能神经元进行性退变、死亡,多巴胺合成或传导能力减弱,导致锥体外系主导的运动调节功能受损是PD的主要病理变化。PD的一个重要病理特征就是α-synuclein突触核蛋白异常聚集形成的路易小体(Lewy body)。此外,MPTP和rotenone等环境毒素的影响造成神经元线粒体的功能障碍,促进氧化应激和有害自由基的生成,也会致使多巴胺能神经元大量变性死亡[7]。目前对PD发病机制的研究主要涉及遗传、年龄老化、环境、免疫炎症、氧化应激、线粒体功能障碍、细胞凋亡等方面。已被证实与PD相关的基因包括α-synuclein(PARK1、PARK4)、Parkin基因(PARK2)、UCH-L1(PARK5)、PINK1(PARK6)、DJ-1(PARK7)、LRRK2(PARK8)、ATP13A2(PARK9)、GBA等数10种[8]。PD 致病基因多样化证实了遗传多态性在PD发病中所起的重要作用。
VPS35是Retromer复合体的最重要元件,它与VPS26和VPS29 组成复合体并与GTP以及Rab7相互作用于EEA[9],在流动性下降的同时募集更多的货物蛋白,继而形成稳定的成核复合体,从endosome向TGN中以囊泡的方式转运高尔基体、内质网、溶酶体、核膜等部位之间的大小不同的颗粒物质[10],保证了许多跨膜的受体蛋白的代谢平衡和重复利用[11]。研究表明VPS35的常染色体显性突变D620N(Asp620Asn)、P316S(Pro316Ser)、R524W(Arg524Trp)等会导致PD发病[12],但具体病理机制尚不明确,目前关于突变体及VPS35表达不足或功能缺失,对于PD的致病机制研究主要集中在α-synuclein 的异常聚集[13]、线粒体功能障碍导致多巴胺能神经元的损失[14]、抑制VPS35与WASH复合体结合[15]、增加MPP+的毒性作用[16]、干扰AMPA 受体的转运、神经元突触成熟障碍[17]等方面。
有研究显示,VPS35缺乏对多巴胺能神经元的损伤尤其严重,并且在临床PD患者的黑质区域也出现了VPS35减少的现象[18]。抑制VPS35的表达会导致新生的海马区神经元树突棘和轴突肿胀而表现出变性样的形态。有趣的是PD的病理特点是这些区域的多巴胺能神经元的变性死亡,而这些域也是DRD1表达最丰富的部位。一些研究证实,参与细胞内外物质转运的蛋白介质如DRiP78[19]、COPI[20]、SNX5[21]等都能与DRD1相互作用并调控其在质膜和细胞表面的运输和成熟分布。在化学性质上,DRD1属于G蛋白偶联(G protein coupled)受体家族的七次跨膜蛋白,其从产生、运输以及最终成熟定位在细胞膜上,时刻处于一个动态的过程。VPS35与GTP以及Rab7相互作用并与早期内体(early endosome)结合[9],由胞内体向高尔基体网状结构逆向转运细胞膜、高尔基体、内质网、溶酶体、核膜等部位之间的货物蛋白,这些蛋白与DRD1的生化性质以及转运特点都高度相似。根据相关报道显示,在VPS35参与转运的货物蛋白中,有些与神经退行性疾病密切相关,如APP[22]、BACE1[23]、AMPA 受体[24]等,作为PD中有重要作用的DRD1,我们猜想VPS35是否也能够调控其转运过程。前期研究结果表明,VPS35虽然对DRD1的基因表达水平影响不大,但能够有效增加DRD1蛋白在细胞膜上的水平,从而增加接受传导多巴胺的受体数量,并且促进内吞入胞的DRD1再循环至细胞膜上,而内吞入胞的DRD1除了再循环返回细胞膜上[6]。在本文降解实验中,我们发现VPS35能够显著促进胞内DRD1通过溶酶体途径降解(见图2)。有趣的是,之前有报道显示VPS35与VPS26和VPS29在EEA的囊泡表面形成复合体,将Cargo蛋白逆向转运至TGN,避免其被运至溶酶体而降解[25],而有些研究结果则表明VPS35促进诸如α-synuclein等蛋白的降解,从而减少神经损伤[26,27]。因此VPS35对DRD1降解调控的具体机制仍需要更深入地探索研究。VPS35的D620N、R524W、P316S等突变已被证实与PD相关,我们前期证实VPS35(D620N c.1858G>A)突变不能有效地调控细胞膜上DRD1的内吞及再循环上膜及在信号转导,说明D620N突变干扰了VPS35的正常生理功能,而VPS35的相关突变是否影响DRD1的降解也有待进一步研究阐明。
本研究证实了VPS35有效通过溶酶体途径调控DRD1的降解,随着对VPS35功能的深入研究,其在以帕金森病和阿尔茨海默病为代表的神经退行性疾病中的相关作用机制也将得到更加详尽的揭示。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
读者(2022年9期)2022-04-22
河南科技(2022年6期)2022-04-22
老年医学研究(2021年5期)2022-01-19
南方医科大学学报(2021年8期)2021-09-10
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
疯狂英语·新读写(2020年3期)2020-06-06
时代英语·高一(2019年5期)2019-09-03
