
运动神经元病合并额颞叶痴呆1例报告并文献复习
2019-11-09 02:19:20刘江峰
中风与神经疾病杂志 2019年10期
刘江峰
运动神经元病(Motor neuron disease,MND)是一组以上和/或下运动神经元损害,主要表现为肌无力和萎缩、延髓麻痹和锥体束征的神经系统变性疾病;额颞叶痴呆(Frontotemporal dementia,FTD)是一组以明显人格、行为改变和语言障碍为特征的痴呆综合征。随着人们对疾病的认识和研究的深入,不断有运动神经元病合并额颞叶痴呆的个案报道,且发现两种疾病可能存在相似的发病机制。为提高对本病的认识,现将我院近期收治的1例运动神经元病合并额颞叶痴呆病例报道如下。
1 临床资料
患者,男性,62岁,因“进行性精神行为异常3 y,言语不清1 y余”于2019年4月22日入院。患者3 y前无明显诱因出现精神行为异常,表现为重复讲一些话、讲他人坏话,打牌时经常多拿或少拿牌,并逐渐出现执行力、理解力、计算力、沟通能力、记忆力减退,常常答非所问,深夜无故外出,强迫性饮食,穿衣服时不能按顺序穿,性格由原来暴躁变得温和,于当地医院就诊,诊断为“老年性痴呆”,未予以特殊治疗。1 y前患者无明显诱因出现言语不清,伴双上肢远端肌肉萎缩,双上肢乏力、持物不稳、饮水呛咳。病程中无头痛、头晕及呕吐、无肢体麻木、无随地大小便等行为。既往体健,无高血压及糖尿病病史;每天吸烟1包,嚼槟榔2包;嗜酒40余年,每天2~3 两,已戒酒1 y。无家族遗传疾病史。
查体:腋温:36 ℃,呼吸:21次/min,脉搏:87次/min,血压:121/95 mmHg。查体欠合作,神清,言语不清,慢性病容,注意力不集中,执行力、计算力、理解力、沟通能力均不同程度减退,记忆力损害较轻,视空间能力尚保留,颈软,双侧瞳孔等大等圆,直径约3 mm,对光反射灵敏,伸舌居中,咽反射正常,舌肌震颤、舌萎缩,双上肢骨间肌、大小鱼际肌、蚓状肌萎缩(见图1),上臂、前臂、肩胛带肌群未见明显萎缩,未见肌束颤动;双上肢近端肌力5级、远端肌力5-级,双下肢肌力5级,四肢肌张力正常,双侧膝反射减弱,余深反射均可引出,霍夫曼征等病理征均(-);深浅感觉正常,共济运动:指鼻试验不配合,跟膝胫试验不配合,闭目难立征(-)。辅助检查:血常规、大便常规+OB、尿常规、心肌酶、肝肾功能、血糖血脂、电解质、凝血常规、输血前4项、脑脊液常规、脑脊液生化、脑脊液细胞学、心电图、胸片、脑电图均正常。MMSE评分:9分。头部MRI+MRA(见图2):脑萎缩(以额颞叶为主),头部MRA未见异常。肌电图:双侧小指展肌、拇短展肌、第一骨间肌、指总伸肌、肱二头肌、胸10椎旁肌、股内肌、胫前肌、腓肠肌、趾短伸肌均可见大量诱发电位、MUP明显宽大,呈单时相或混合期,结合神经速度提示:呈广泛性神经源性损害,支持运动神经元病诊断。
该患者病程3 y,双上肢远端肌萎缩、肌无力,构音障碍、舌肌萎缩、饮水呛咳、强哭强笑,肌电图提示广泛性神经源性损害,根据运动神经元病El Escorial[1]诊断标准,可拟诊为运动神经元病。此外,患者存在精神行为异常、认知功能减退,性格改变,饮食改变,执行力、理解力、计算力、沟通能力减退,记忆力损害较轻,视空间能力尚保留,MMSE评分低于正常,头部MRI提示额颞叶萎缩,根据Neary等[2]制定的诊断标准,可临床诊断为额颞叶痴呆,确诊需行活检或死后尸检。故本例患者可诊断为运动神经元病-额颞叶痴呆综合征。
2 讨 论
运动神经元病(MND)是一组主要累及锥体束、脑干运动神经核、脊髓前角细胞的神经退行性疾病,通常在症状出现后3~5 y内死亡,约90%的MND病例呈散发性,10%的MND为家族性。近年来,越来越多人认为MND是一种可伴有认知障碍和行为改变的多系统疾病,约10%~15%的MND患者符合额颞叶痴呆的诊断标准[3],FTD症状可在运动症状之前、之后或同时出现。以下就MND-FTD的遗传和病理机制、临床特征、治疗及预后进行讨论。
2.1 发病机制 MND-FTD目前尚无确切发病机制,已有研究发现某些基因突变可引起MND,如铜/锌超氧化物歧化酶(Cu/Zn superoxide dismutase 1,SOD1)基因突变、FUS基因突变、TARDBP基因突变等[4];而MAPT基因突变、GRN基因突变等[5]可引起家族性额颞叶痴呆,但这些突变基因都不能解释MND-FTD。运动神经元病、额颞叶痴呆共病患者越来越多,这提示两者可能存在共同的发病机制。DeJesus-Hernandez M和Renton AE等人发现C9orf72基因第一个内含子的六核苷酸GGGGCC扩增是家族性运动神经元病和额颞叶痴呆最主要的遗传原因[6,7]。在家族性MND-FTD患者中,多达50%~70%的患者携带C9orf72基因重复扩增[8]。C9orf72基因重复扩增原因尚不明确,目前认为DNA高甲基化可能是C9orf72运动神经元病-额颞叶痴呆疾病的一个主要致病机制[9]。C9orf72基因重复扩增致病可能通过以下3种机制[10,11]:(1)双向转录产生包含G4C2的RNA和G2C4的反义RNA,能够隔离蛋白质,从而干扰其生理功能;(2)非AUG启动子重复启动翻译产生二肽重复蛋白质(DPR),DPR的形成和累积可产生细胞毒性;(3)C9orf72基因外显子序列转录减少可引起其单倍体功能不全。此外,有文献报道,在少数MND-FTD患者或家族中发现了VCP、UBQLN2、OPTN和SQSTM1基因突变[12],这说明家族性MND-FTD存在着多种致病基因遗传(即寡基因遗传)。
神经元胞质异常蛋白的聚集是大多数神经退行性疾病的重要特征,有研究发现,约40%~50%的额颞叶痴呆、75%~90%的运动神经元病患者存在TDP-43病理包涵体[8],TDP-43异常蛋白质沉积是FTLD-TDP、MND的病理特征。Mackenzie IR等人根据TDP-43病理形态及分布的不同,将FTLD-TDP分为以下4种亚型[13,14]:A型:以新月形或椭圆形神经元胞质内含物(NCI)和多而短的营养不良性神经元突起(DN)为特征,主要位于上皮质,该亚型与GRN基因突变相关;B型:NCI数量中等,DN数量很少,分布于整个皮质层,MND-FTD及C9orf72基因突变与B型关联最强;C型:NCI数量很少,DN数量多且长,主要分布于上皮质;D型:以神经元核内包涵体、短DN为主,NCI数量少,主要位于上皮质,该亚型与VCP基因突变临床表型具有独特相关性。除此之外,额颞叶痴呆变性(FTLD)还与tau蛋白、肉瘤融合蛋白(FUS)等有关[5]。运动神经元病的TDP-43蛋白在脑内的沉积模式与额颞叶痴呆不同,Brettschneider J[15]等人根据pTDP-43病理蛋白分布及解剖,提出MND病理分期:第一阶段:pTDP-43蛋白主要累及运动皮质、脑干运动神经核及脊髓运动神经元等远动系统;第二阶段:累及额中回、脑干网状核、小脑前核、红核;第三阶段:累及额叶前部、皮质感觉区及纹状体;第四阶段:累及颞叶内侧及海马。这表明MND是一种既选择性影响运动系统、又影响其他神经元和胶质细胞的多系统神经退行性TDP-43蛋白病[14]。但是否所有的运动神经元病病理过程都以同样方式进展,目前尚不清楚。
2.2 临床特征 MND是一种进行性成人发病的神经退行性疾病,疾病类型主要包括肌萎缩侧索硬化(ALS)、进行性延髓麻痹(PBP)、进行性肌萎缩(PMA)和原发性侧索硬化(PLS),ALS是最常见的MND类型,PBP、PMA、PLS最终可进展为ALS。MND因疾病类型不同,而表现出不同的临床症状组合,主要表现:构音障碍、吞咽困难、饮水呛咳、流涎、肢体萎缩和无力、肌束颤动、呼吸困难等,以及病理征阳性、腱反射亢进、强哭强笑、肌张力增高等体征。FTD是一种具有明显临床表型的异质性疾病,其类型主要包括行为异常型额颞叶痴呆(bvFTD)、语义性痴呆(SD)、进行性非流利性失语(PNFA)。bvFTD主要表现在行为、个性、情感、执行控制力等方面的变化,这些变化包括抑制解除、冷漠、缺乏同情心、强迫/刻板行为、饮食变化等,bvFTD行为症状改变主要与额叶内侧、额岛叶皮质、扣带回前部功能障碍有关[5];SD以单字理解障碍和命名性失语为主要特征,患者言语流利、语法正确,但不能理解单词含义、找词困难,SD主要与优势半球颞叶受累有关[5]。PNFA主要表现为语言表达及交流能力下降,找词困难,言语减少,理解力、记忆及视空间能力通常保留,PNFA与优势半球额下回Broca区及岛叶前部受累有关[5]。
MND的临床症状可发生于FTD患者,同时MND在病程中也可表现语言或行为损害症状。Long Z[16]等发现大部分MND-FTD患者有语言障碍,且具有高度普遍及异质性等特点,其语言障碍包括失语症、单字理解障碍、句子理解障碍、语法缺陷,MND-FTD语言障碍严重程度与FTD的语言表型相似。一项涉及279名散发性MND/FTD患者的研究中[17],约50%的MND患者存在认知障碍,10%~15%患者符合FTD标准,且主要表现为bvFTD。Cortes-Vicente E[18]等人在22例MND-bvFTD患者随访期间发现选择性上肢远端肌无力和萎缩伴不明显下肢无力是主要的运动损害特征,严重的吞咽困难和吸入性肺炎是最常见的死亡原因,没有痴呆的患者多数死于呼吸衰竭,约1/3患者一生中只表现为单纯的下运动神经元(LMN)症状,从未出现上运动神经元(UMN)症状,但患者行基因检测或死后接受神经病理检查,却支持MND诊断。
2.3 治疗及预后 目前,暂无特效方法治疗MND-FTD,虽然利鲁唑在MND治疗效果已被证实,但目前没有研究直接检测利鲁唑在MND-FTD中的作用;FDA批准治疗阿尔茨海默病的药物对FTD也没有获益,且有研究表明乙酰胆碱酯酶抑制剂可能会加重FTD症状[19],美金刚在FTD的治疗效果尚不明确。对于伴有躁动、易激惹、攻击性行为的MND-FTD患者使用抗精神病药物可能有用[8]。MND-FTD具有高致死性,预后差,有研究表明,MND-FTD患者疾病进展比没有痴呆的MND进展更快[3],年龄大、延髓起病、存在认知功能损害、教育程度低的患者生存时间更短、预后更差[8,17]。
2.4 小结 综上,虽然MND-FTD在遗传和病理方面取得较大进展,但是否所有MND或FTD患者均有进展为MND-FTD的风险,或存在某些易感因素导致一些患者更易患MND-FTD,目前尚不明确。因MND-FTD的高致死率及致残率,目前迫切需要针对该疾病基本机制的有效治疗方法,针对C9orf72基因转录和翻译等方面进行干扰可能是MND-FTD的治疗靶点之一,但C9orf72基因及其他致病基因的发生和发展机制有待进一步阐明。目前有实验对C9orf72基因转录及翻译等方面进行研究,但尚未取得明显进展。MND-FTD目前无统一的诊断标准,多数人采用同时符合MND和FTD各自标准来诊断这一疾病。未来,对MND-FTD致病机制及诊断标准进行深入研究将有利于治疗和预后,为MND-FTD患者的有效治疗带来曙光。
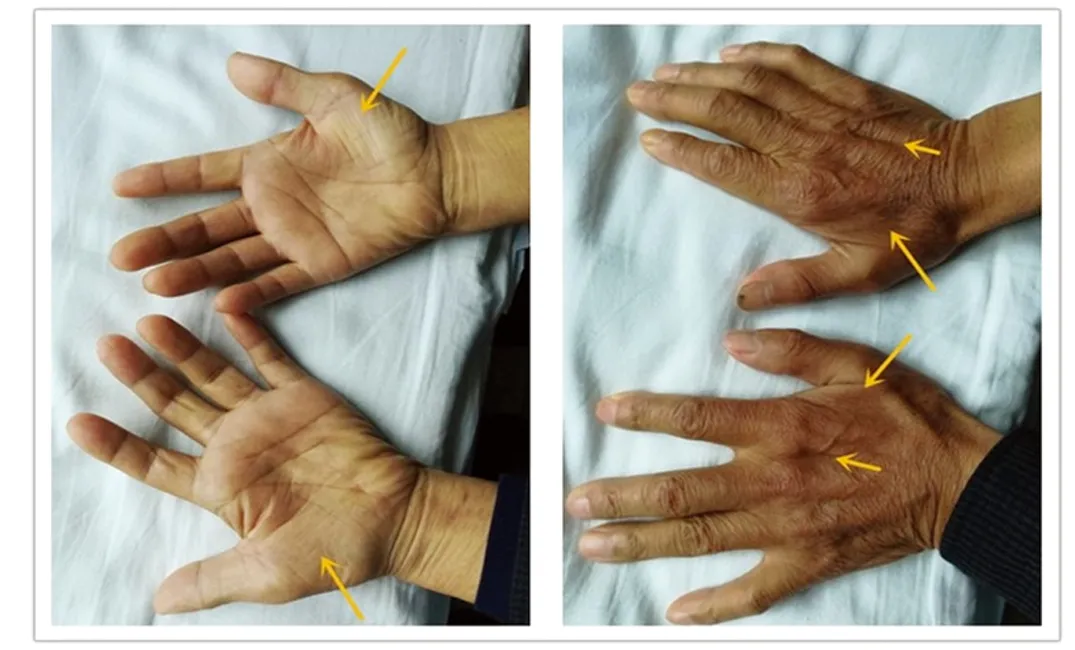
图1 患者双侧骨间肌、大小鱼际肌、蚓状肌萎缩
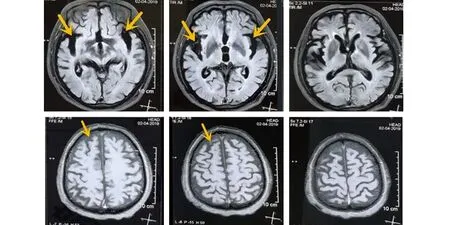
图2 双侧额颞叶萎缩,以颞叶萎缩明显
猜你喜欢
考试与评价·高二版(2020年2期)2020-09-10 07:22:44
家庭科学·新健康(2019年9期)2019-10-21 03:55:48
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
中外医疗(2015年16期)2016-01-04 06:51:42
实用手外科杂志(2015年1期)2015-08-27 01:52:06
医学研究杂志(2015年9期)2015-07-01 17:28:00
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
中医研究(2013年9期)2013-03-11 20:27:37
中国医学科学院学报(2013年3期)2013-03-11 20:25:53
河北医科大学学报(2010年10期)2010-03-25 10:14:57
