
Au 改性异构化原料预加氢催化剂
2019-11-08 00:48张孔远王倩倩崔程鑫张朋伟刘晨光
石油化工 2019年10期
张孔远,王倩倩,崔程鑫,张朋伟,刘晨光
(中国石油大学(华东) 化学工程学院 重质油国家重点实验室,山东 青岛 266555)
C5/C6烷烃异构化可有效地提高产品的辛烷值,且不增加烯烃和芳烃的含量,异构化产物是优良的汽油调和组分[1-2]。异构化工艺作为一种环境友好的炼油技术,已成为清洁汽油加工的必要工艺之一[3]。国外异构化装置以美国UOP 公司和法国Axens 公司的异构化技术为主,国内多家公司也相继开发成功了多种C5/C6异构化技术并进行了工业应用[4-5]。工业C5/C6异构化原料主要来源于原油常压蒸馏的拔头油和催化重整的抽余油,含有一定量的烯烃和苯,在临氢异构化过程中易加氢饱和放出大量的热[5]。反应温度升高一方面抑制了异构化反应,降低C5/C6烷烃异构化反应的平衡转化率,并加快裂解反应速率,造成异构化产物液体收率下降[6];另一方面促进烃类的生焦,造成催化剂活性降低甚至失活。因此,在异构化反应前对原料进行预加氢,再将原料中的烯烃和芳烃饱和加氢,可获得更加平稳的操作条件,提高异构化反应的转化率[7]。
本工作选取工业Al2O3载体,采用等体积浸渍法制备Pt/Al2O3和Pt-Au/Al2O3系列催化剂,并进行了H2-TPR,HRTEM,XPS,XRD 等表征;分别采用含2%(w)苯的正己烷和含2%(w)苯、1%(w)1-己烯的正己烷溶液为模型化合物,在小型固定床加氢反应器上对催化剂进行了加氢活性评价。
1 实验部分
1.1 主要试剂
AuCl3·HCl·4H2O,H2PtCl6·6H2O,C6H6,C6H14:分析纯,国药集团化学试剂有限公司;HCl:36%(w)~38%(w),西陇科学股份有限公司;氢气:100%(φ),青岛天源气体制造有限公司;Al2O3载体:工业品,直径1.6 mm,长度2 ~3 mm。
1.2 催化剂的制备
称取一定量的载体,根据载体吸水率采用H2PtCl6·6H2O 溶液浸渍载体,浸渍后静置12 h,经干燥和焙烧,制得Pt 负载量为0.3%(w)的新鲜Pt/Al2O3催化剂。
称取一定量的载体,根据载体吸水率采用AuCl3·HCl·4H2O和H2PtCl6·6H2O共浸液浸渍载体,浸渍后静置12 h,经干燥和焙烧,制得Pt 负载量均为0.3%(w)、Au 含量(w)分别为0.02%,0.05%,0.10%,0.20%的Pt-Au/Al2O3系列催化剂,分别记为Au-1,Au-2,Au-3,Au-4。
1.3 催化剂的表征
采用荷兰PANalytical 公司X’PERT PRO MPD型X 射线衍射仪进行XRD 表征;采用日本日立公司JEM-2100UHR 型高分辨率透射电子显微镜对催化剂表面Pt 粒子和Au 粒子进行HRTEM 表征;采用Quantachrome 公司CHEMBET-3000 型TPD/TPR 分析仪对试样进行H2-TPR 表征;采用赛默飞世尔公司ESCALab250 Xi 型X 射线光电子能谱仪对催化剂Pt 粒子和Au 粒子的化学状态进行XPS表征。
1.4 催化剂的活性评价
催化剂加氢活性评价实验分别采用含2%(w)苯的正己烷(记为原料Y1)和含2%(w)苯、1%(w)1-己烯的正己烷溶液(记为原料Y2)为模型化合物,在10 mL 固定床反应器上(内径1.0 cm,长约80 cm)进行,催化剂装填量为5 mL,位于床层中间,两侧用石英砂充填。催化剂还原条件为:压力0.5 MPa,温度350 ℃,氢气流量100 mL/min。催化剂评价条件为:温度为45 ℃时,压力1.5 ~2.0 MPa,氢油体积比400 ~600,液态空速2 ~8 h-1。稳定2 h 后,取两个平行样,采用美国Agilent 公司GL 6820 型气相色谱仪对原料及产物进行PONA分析。
2 结果与讨论
2.1 H2-TPR 表征结果
图1 为催化剂的H2-TPR 曲线。由图1 可知,Pt/Al2O3催化剂的还原峰在255 ℃,Au/Al2O3催化剂的还原峰在275 ℃,Pt/Al2O3催化剂中加入Au后,未单独出现Au 的还原峰。试样Au-1 的还原峰位于300 ℃,随着Pt-Au/Al2O3各催化剂试样中Au含量的增加,耗氢峰向低温方向移动,这可能是由于Au 加入后,随着Au 含量的增加,Pt 或Pt-Au合金的粒子增大,造成还原温度降低;但Pt-Au/Al2O3系列催化剂试样的还原峰温度始终高于Pt/Al2O3催化剂和Au/Al2O3催化剂,这是Pt 和Au 相互作用或形成了合金的结果。
2.2 HRTEM 表征结果
图2 为Pt/Al2O3催化剂和Au-2 催化剂试样的HRTEM 照片及快速傅里叶变换(FTT)图像。由图2 可知,随着Au 的加入,催化剂中的Pt 晶粒明显增大。由图2b 可知,测量得晶面间距为0.227 nm,属于Pt(111)晶面(JCPDS 4-0802)。由图2d 可知,测量得晶面间距为0.229 nm,介于Au(111)晶面的晶面间距0.236 nm(JCPDS 4-0784)和Pt(111)晶面的晶面间距0.227 nm 之间,可归于Pt-Au(111)晶面。证明了在Pt/Al2O3催化剂中引入Au 后形成了Pt-Au 合金,即Au 的加入稀释了催化剂表面Pt 粒子。

图2 Pt/Al2O3 催化剂和Au-2 催化剂试样的HRTEM 照片及FFT 图像Fig.2 HRTEM images and fast Fourier transformation(FFT) images of Pt/Al2O3 catalysts and Au-2 catalyst samples.
2.3 XPS 表征结果
实验制备的Pt-Au/Al2O3催化剂中Pt 和Au 含量低,不在XPS 表征的灵敏度范围内,因此制备了Pt 负载量为5%(w)的Pt/Al2O3催化剂和Pt 负载量为5%(w)、Au 含量为0.83%(w)的Pt-Au/Al2O3催化剂。图3 为催化剂的XPS 谱图。由图3 可知,Pt 4f 与Al 2p 谱峰叠合,但可进行定性分析,Pt/Al2O3和Pt-Au/Al2O3的Pt 4f 与Al 2p 的 电 子 结 合能分别为74.65 eV 和74.55 eV,采用XPSpeak 软件分峰处理可得Pt 4f 峰值分别为74.90 eV 和74.95 eV,Pt 4d 电子结合能分别为316.20 eV 和316.33 eV,Pt 主要以+4 价的氧化态存在,这是因为在催化剂干燥焙烧的过程中氯铂酸分解生成Pt 的氧化物[8-9]。零价Au 的4f7/2的电子结合能为83.8 eV,+3 价Au 的4f7/2的电子结合能为87.5 eV。由图3c可知,催化剂的Au 4f 峰出现在83.94 eV 和87.55 eV 处,即可能存在两种价态的Au,+3 价和零价,而零价Au 的4f5/2峰出现在87.6 eV,与+3 价Au的4f7/2峰接近,二者可能存在叠合,结合H2-TPR表征结果可知,催化剂表面存在可还原的Au,可见Au 以+3 价和零价两种状态存在。在添加Au 前后Pt 均为+4 价,但Au 的添加使Pt 4d 的电子结合能产生了0.05 eV 的化学位移,使Pt 4f 的电子结合能产生了0.13 eV 的化学位移,可能的原因是Au 的加入使Pt 周围的化学环境发生了轻微变化,但并未对Pt 的电子性质产生明显影响。
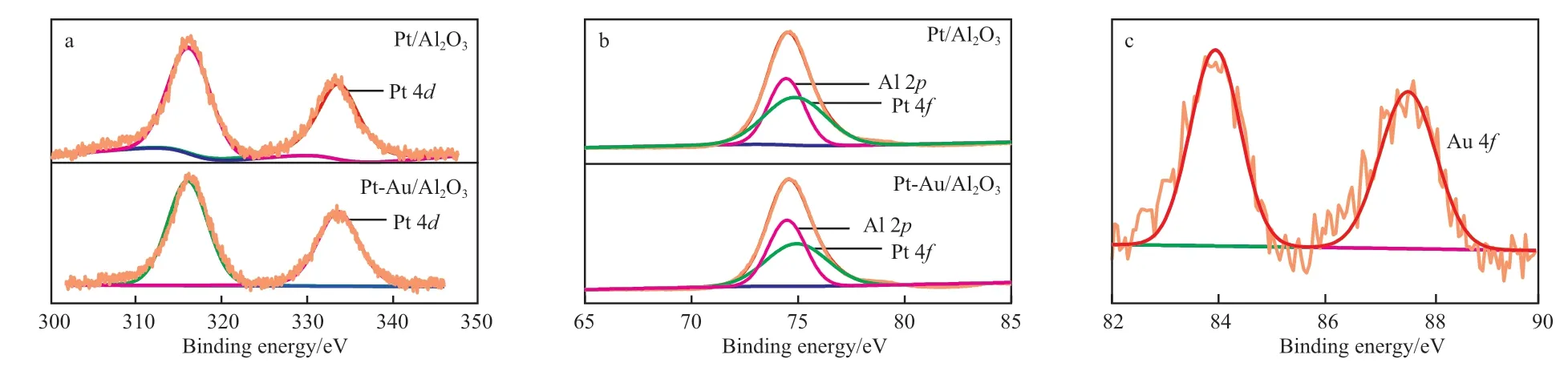
图3 催化剂的XPS 谱图Fig.3 The XPS spectra of catalysts.
2.4 XRD 表征结果
图4 为Al2O3载体、Pt/Al2O3催化剂和Pt- Au/Al2O3催化剂的XRD 谱图。由图4 可知,Al2O3载体的出峰位置在2θ=36.8°,39.1°,46.2°,66.7°处,为γ-Al2O3的特征峰;Pt/Al2O3催化剂的出峰位置主要在2θ=37.5°,39.8°,46.1°,66.5°处,Pt(111)晶面的衍射峰位置为39.8°,Pt(200)晶面的为46.2°,Pt(220)晶面的为67.4°,与γ-Al2O3的衍射峰位置十分接近,由于γ-Al2O3有较大的比表面积,Pt 分散较好,二者的衍射峰易重合,因此Pt/Al2O3催化剂上未能看出单独的Pt 衍射峰。Pt-Au/Al2O3催化剂的出峰位置在2θ=38.2°,39.6°,44.5°,46.0°,64.6°,66.6°处,其中38.2°为Au(111)晶面的衍射峰,44.5°为Au(200)晶面的衍射峰,64.6°为Au(220)晶面的衍射峰,即出现明显的Au 的晶体衍射峰;而39.6°,46.0°,66.6°处的衍射峰与Pt/Al2O3催化剂相比,均稍左移,但偏移很小,基本上无影响,因此Au 的添加对催化剂中Pt 的晶相无显著影响。
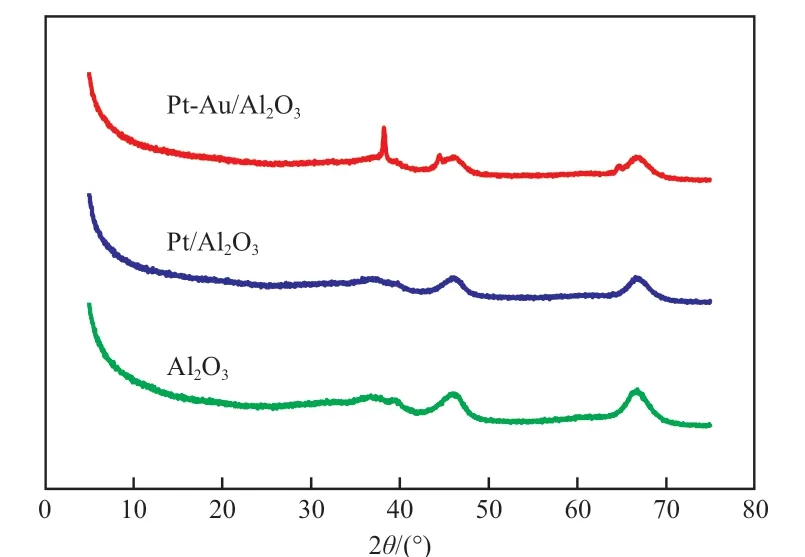
图4 Al2O3 载体、Pt/Al2O3 催化剂和Pt-Au/Al2O3 催化剂的XRD 谱图Fig.4 XRD patterns of Al2O3 carrier,Pt/Al2O3 and Pt-Au/Al2O3 catalysts.
2.5 催化剂加氢活性的评价结果
图5 为不同Au 含量Pt-Au/Al2O3催化剂的加氢活性曲线。由图5 可知,苯生成环己烷反应与1-己烯生成正己烷反应在评价过程中始终保持100%的选择性。催化剂的加氢活性随着Au 含量的增加先升高后降低,这是由于当Au 的负载量较低时,只有少量的Pt 与Au 形成Pt-Au 合金,分散在大比表面积氧化铝上的Pt 粒子具有较高的分散度,相对粒径较小,小晶粒Pt 的苯加氢活性低;随着Au 含量的增加,生成的Pt-Au 合金逐渐增加,当Au 含量与Pt 达到最佳比例时Pt-Au 合金量达到最高,还原后的的Pt 粒子大小适宜于苯加氢,催化剂的苯加氢活性最高;进一步添加Au 时,Au 与Pt 形成合金的粒径进一步增加,还会对Pt 粒子表面进行覆盖和包裹,阻碍Pt 粒子活性位对反应物的吸附,使催化剂苯加氢活性降低。Au 添加量为0.05%(w)时,催化剂的苯加氢转化率最高,对于Y1 原料,苯的转化率达到96.50%,产物环己烷的选择性为100%。对比Y1 和Y2 原料的转化率曲线可知,在原料中加入1-己烯后,苯的加氢转化率大幅度降低,这是由于原料中加入烯烃后,苯加氢反应与1-己烯加氢反应为竞争反应,烯烃比芳烃更容易吸附在加氢活性位上,使苯可吸附的活性位减少,苯加氢活性降低;对于Y2 原料,当Au 为最佳含量时,苯的转化率为33.33%,产物环己烷选择性为100%,1-己烯的转化率为100%,产物正己烷选择性为100%。因此,保持Pt 负载量为0.3%(w)不变,当Au 含量为0.05%(w)时,Pt-Au 合金形成最佳比例,Pt-Au/Al2O3催化剂的加氢活性最高。
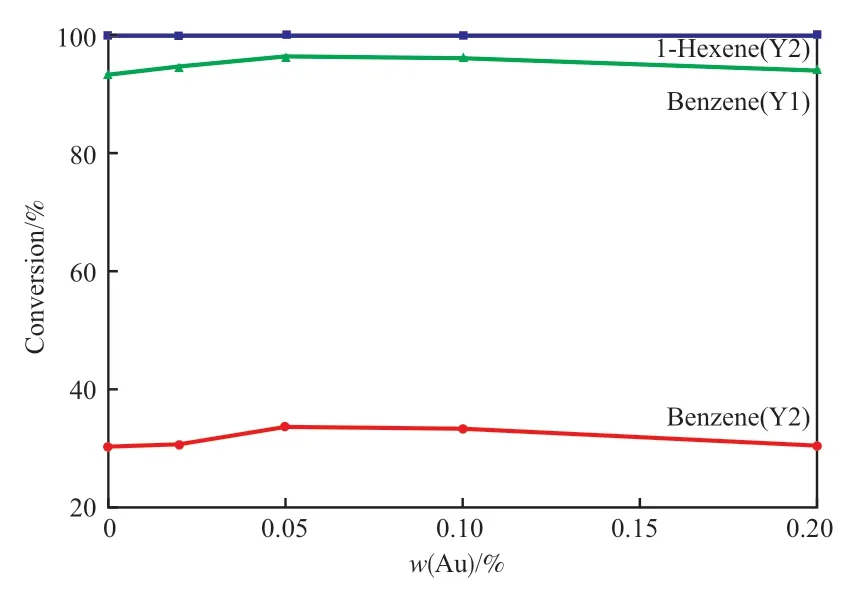
图5 Pt-Au/Al2O3 催化剂的加氢活性曲线Fig.5 Hydrogenation activity curves of Pt-Au/A12O3 catalysts.
3 结论
1)Pt/Al2O3加氢催化剂表面添加Au 后,Au以+3 价和零价两种状态存在,Au 粒子与Pt 粒子形成Pt-Au 合金,改变了Pt 周围的化学环境,但并未改变Pt 的价态和晶相结构。
2)保持Pt 负载量为0.3%(w)不变,当Au含量为0.05%(w)时,Pt-Au 合金形成最佳比例,催化剂的加氢活性最高。
3)原料中1-己烯的加入会显著降低芳烃的加氢转化率,1-己烯和苯在Pt-Au/Al2O3催化剂表面存在竞争吸附,二者的加氢反应为竞争反应。
猜你喜欢
化工管理(2021年3期)2021-01-29
石油石化绿色低碳(2020年2期)2020-12-31
化工管理(2020年26期)2020-10-09
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
石油化工技术与经济(2019年3期)2019-02-13
石油石化绿色低碳(2019年6期)2019-01-14
西安工业大学学报(2018年6期)2018-02-13
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
应用化工(2015年3期)2015-04-01
