
Cre-loxP重组酶技术敲除酿酒酵母丙酮酸脱羧酶编码基因pdc1和pdc5
2019-09-27 02:26刘磊王长丽葛菁萍
生物技术通讯 2019年4期
刘磊,王长丽,葛菁萍
黑龙江大学a. 农业微生物技术教育部工程研究中心,黑龙江 哈尔滨150500;b. 生命科学学院微生物省高校重点实验室,黑龙江 哈尔滨150080
2,3-丁二醇(2,3-butanediol;2,3-BD)及其衍生物是重要的平台化合物,在化工、燃料、食品等领域具有很高的应用价值[1-2]。自然界中具有较强2,3-BD 生产能力的微生物大多为条件致病菌,如克雷伯菌属与肠杆菌属,大规模工业化生产2,3-BD 会受到限制[3]。与这些条件致病菌相比,酿酒酵母作为公认安全(generally recognized as safe,GRAS)微生物,被广泛用于各种化学品和燃料的工业化生产中[4-5]。酿酒酵母是真核模式生物,其遗传背景清晰,研究较为透彻。虽然野生型酿酒酵母产2,3-BD 的量较少,但可以利用基因工程方法对其进行改造[6]。酿酒酵母发酵的主产物是乙醇,故在其代谢途径中,丙酮酸脱羧酶(pyruvate decarboxylases,PDC)至关重要。PDC是一种非氧化酶,位于细胞质基质中,可以催化丙酮酸脱羧,首先生成乙醛和CO2,乙醛在相关酶的催化下进一步生成乙酸和乙醇[7]。因此,可以通过构建含有敲除组件的质粒,敲除酿酒酵母中丙酮酸脱羧酶的编码基因(pdc1、pdc5和pdc6)来阻断部分副产物生成,以提高2,3-BD 的产量[8-9]。
基因敲除技术普遍应用于微生物代谢工程及菌种改造等领域,其中Cre-loxP 重组酶技术可以特异性控制Cre 重组酶在特定组织的表达,以实现在特定组织细胞中对目的基因的敲除或改造[10]。Cre-loxP 重组酶技术系统由Cre 重组酶与特异性靶位点loxP 组成。Cre 重组酶是位点特异性重组酶,通过识别特异的DNA 序列(loxP 位点),可以敲除loxP 位点间的基因序列[11],同时又能敲除标记基因kanMX,有效解决了在同一株酿酒酵母细胞中进行多基因敲除时抗性筛选标记选择的难题[12]。应用Cre-loxP 重组酶技术,首先需要选取pUG 系列质粒,因其上2 个loxP 位点中含有筛选标记基因(如loxP-kanMX-loxP),同时需要构建基因敲除组件,使loxP-kanMX-loxP两端含有40~45 bp 敲除基因的同源片段,并将其导入受体细胞,即可实现目的基因的敲除,若向其中导入pSH 系列质粒(可表达Cre 重组酶),又能将标记基因消除[13-14]。
我们以质粒pUG6 基因组DNA 为模板,克隆出两端含40 bp 与酿酒酵母pdc1、pdc5同源的线性片段pdc1-loxP-kanMX(1693 bp)和pdc5-loxP-kanMX(1672 bp),以遗传霉素G418 为抗性筛选标记,构建含有pdc1、pdc5同源序列loxP-kanMX-loxP的质粒,将构建后的质粒转化酿酒酵母H5 进行筛选及鉴定,并通过摇瓶发酵试验测定菌株发酵产量变化情况。本研究分别敲除了酿酒酵母体内丙酮酸脱羧酶基因pdc1、pdc5,为进一步连续敲除pdc1、pdc5奠定了实验基础。
1 材料与方法
1.1 材料
酿酒酵母H5 和大肠杆菌DH5α由黑龙江大学微生物重点实验室提供;质粒pMD18-T 购自大连宝生物工程有限公司,pEASY-T3 购自北京全式金有限公司,pUG6 由广西大学杜丽琴教授馈赠;限制性内切酶HindⅢ、BamHⅠ、EcoRⅠ购自北京天玺瑞至有限公司;PfuDNA 聚合酶、TaqDNA 聚合酶购自TaKaRa 公司;TIANgel Midi Purification Kit 普通琼脂糖凝胶DNA 回收试剂盒购自天根生化科技有限公司;BioSpin Gel Extraction Kit 购自杭州博日科技有限公司。
发酵培养基:葡萄糖80 g/L、蛋白胨20 g/L、YNB 3.4 g/L、KH2PO43.4 g/L、K2HPO43 g/L、(NH4)2SO410 g/L,108℃灭菌20 min;LB 培养基:蛋白胨10 g/L、NaCl 5 g/L、酵母浸出物5 g/L,121℃灭菌15 min;YPD 培养基:酵母提取物10 g/L、蛋白胨20 g/L、葡萄糖20 g/L,108℃灭菌20 min。
1.2 引物设计
依据NCBI 中酿酒酵母S288C(序列号:NC_001144.5)的pdc1、pdc5和pUG6 质粒序列(序列号:AF298793.1)设计引物对pdc1-S1 和pdc1-S2、pdc5-S1 和pdc5-S2(表1),用来扩增pdc1、pdc5。引物两端各40 bppdc1、pdc5同源序列(下划线部分),其他20 bp 用于扩增loxP-kanMX-loxP。

表1 引物序列

图1 转化子的PCR 验证示意图
1.3 两端各含40bppdc1、pdc5同源序列的loxP-kanMX-loxP片段的克隆及转化
将复苏后含有pUG6 的大肠杆菌DH5α转接到氨苄青霉素终浓度为100 μg/mL 的LB 液体培养基中,振荡培养至对数生长期,提取质粒DNA,以获得的pUG6 质 粒DNA为模板,pdc1-S1 和pdc1-S2、pdc5-S1 和pdc5-S2 为引物,PCR 扩增目的片段(反应程序:94℃ 5 min;94℃ 30 s,56℃30 s,72℃4 min,35 个循环;72℃10 min)。
PCR 结束后,向反应体系中加入0.25 μLTaqDNA 聚合酶(5 U/μL),进行加“A”尾,72℃水浴中反应10 min,1%琼脂糖凝胶电泳检测PCR产物;用BioSpin Gel Extraction Kit 纯化目的片段,并在16℃金属浴中与pMD18-T 载体过夜连接;将连接产物用热激法转化大肠杆菌DH5α感受态细胞,适当稀释涂布培养至LB 培养基中;随机挑取LB 培养基中的白色单菌落,接种于含氨苄青霉素的LB 液体培养基中,37℃、160 r/min 振荡过夜培养,PCR 验证大肠杆菌转化子;提取重组克隆质粒,BamHⅠ单酶切验证阳性质粒,送上海生工生物工程有限公司测序,用生物学软件分析比对测序结果,验证正确含有目的序列的克隆质粒命名为pTWCL-PDC1 与pTWCL-PDC5,同源重组片段命名为pdc1-loxP-kanMX与pdc5-loxP-kanMX。将验证正确的克隆质粒用醋酸锂转化法分别转化酿酒酵母H5。
1.4 缺失菌株的筛选与鉴定
片 段pdc1-loxP-kanMX与pdc5-loxP-kanMX分别转化菌株酿酒酵母H5 后,设计引物pdc1-A1、pdc1-B1、pdc1-C1、pdc1-D1 与pdc5-A5、pdc5-B1、pdc5-C1、pdc5-D5 进 行PCR 验 证,随机挑取在YPD+G418 平板上生长的单菌落,提取基因组DNA,以基因组DNA 为模板,用引物对pdc1-B1 和pdc1-C1、pdc1-A1 和pdc1-B1、pdc1-A1 和pdc1-D1、pdc1-C1 和pdc1-D1分别对pdc1-loxP-kanMX进行PCR 验证(图1A),用引物对pdc5-B1 和pdc5-C1、pdc5-A5 和pdc5-D5 分 别对pdc5-loxP-kanMX进行PCR 验证(图1B)。
1.5 G418抗性的消除
pSH69 质粒自身带有潮霉素(HyB)标记基因,添加半乳糖可诱导表达Cre 重组酶消除其G418 抗性。用醋酸锂转化法将pSH69 质粒转化重组菌株,将验证成功的转化子接种于液体YPD中,过夜培养后添加终浓度为1%的半乳糖溶液诱导表达Cre 重组酶,继续培养至菌液D600nm约为1.3,稀释涂布于YPD 与YPD+G418 平板上,挑取在YPD+G418 培养基上生长受抑制而在YPD 平板上长势良好的菌落,以基因组DNA 为模板,用引物对pdc1-A1 和pdc1-D1、pdc1-B1 和pdc1-C1 进行PCR 扩增验证抗性片段kanMX是否敲除成功。之后将添加pSH69 质粒的重组菌株涂布于YPD+HyB 平板上培养2~3 d,挑取平板上的单菌落,用灭菌枪头将其分别点殖于YPD+HyB 和YPD 平板上,通过连续传代获得失去HyB 抗性的单菌落。将质粒丢失且pdc1、pdc5分别敲除的菌株命名为H5-01 和H5-02。
1.6 发酵性能检测
将处于对数生长期的酿酒酵母H5、H5-01、H5-02 菌株分别以5%的接种量接种于150 mL/500 mL 的发酵培养基中,于30℃、150 r/min 发酵72 h,每 隔12h取样测 定D600nm和pH 值,通 过HPLC 检测发酵液中乙醇和2,3-BD 浓度的变化。
2 结果
2.1 两端各含40bppdc1、pdc5同源序列的loxP-kanMX-loxP片段的克隆
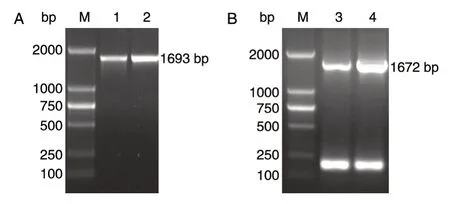
图2 PCR 扩 增loxP-kanMX-loxP 片段
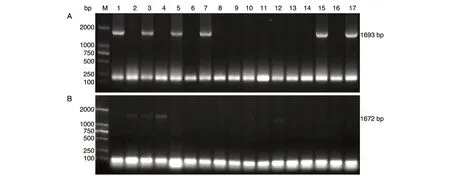
图3 阳性克隆子的菌液PCR 验证
PCR 验证含有质粒pTWCL-PDC1 与pTWCLPDC5 的阳性克隆子,从图2A 可以看出,PCR 产物pdc1-loxP-kanMX片段与目的片段大小相同,为1693 bp,与预期结果相符;从图2B 可以看出,PCR 产物pdc5-loxP-kanMX(1672 bp)片段与预期片段大小相符。
片段pdc1-loxp-kanMX、pdc5-loxP-kanMX纯化后分别连接到克隆载体pMD18-T、pEASY-T3 上,并转化大肠杆菌DH5α,从每种转化平板上随机挑取17 个白色菌落进行菌液PCR 鉴定。图3A 中泳道1、3、5、7、15、17 号阳性克隆子扩增得到的片段均与pdc1-loxP-kanMX大小相符(1693 bp),图3B 中泳道1、2、3、4、5、12 号阳性克隆子扩增得到的片段均与pdc5-loxP-kanMX片段大小相符(1672 bp),说明目的基因pdc1-loxP-kanMX与质粒pMD18-T连接成功,pdc5-loxP-kanMX与质粒pEASY-T3 连接成功。从转化后的质粒pMD18-T、pEASY-T3 中选取阳性克隆子提取质粒,分别经BamHⅠ、EcoRⅠ单酶切验证,将目的片段测序后与NCBI 数据库比对,发现pdc1-loxP-kanMX、pdc5-loxP-kanMX核苷酸序列与NCBI 中公开的基因序列相似度达100%,未发生碱基突变。
2.2 缺失菌株的筛选与鉴定
pdc1、pdc5基因敲除转化子验证分别见图4、5,在B1-C1(732 bp)、A1-B1(1202 bp)、A1-D1(1732 bp)、C1-D1(1066 bp)与A5-D5(1185 bp)、B1-C1(732 bp)处均有清晰条带出现,与预期片段大小相符。

图4 用引物B1-C1(A)、A1-B1(B)、A1-D1(C)、C1-D1(D)的PCR 扩增验证

图5 用引物A5-D5(A)、B1-C1(B)的PCR 扩增验证
2.3 G418抗性的消除
用引物pdc1-A1 和pdc1-D1、pdc1-B1 和pdc1-C1 进行PCR 扩增,验证抗性片段kanMX的结果如图6,在A1-D1(262 bp)处有清晰条带出现,而B1-C1(732 bp)处未扩增出条带,说明kanMX片段已消除,并敲除了pdc1、pdc5。
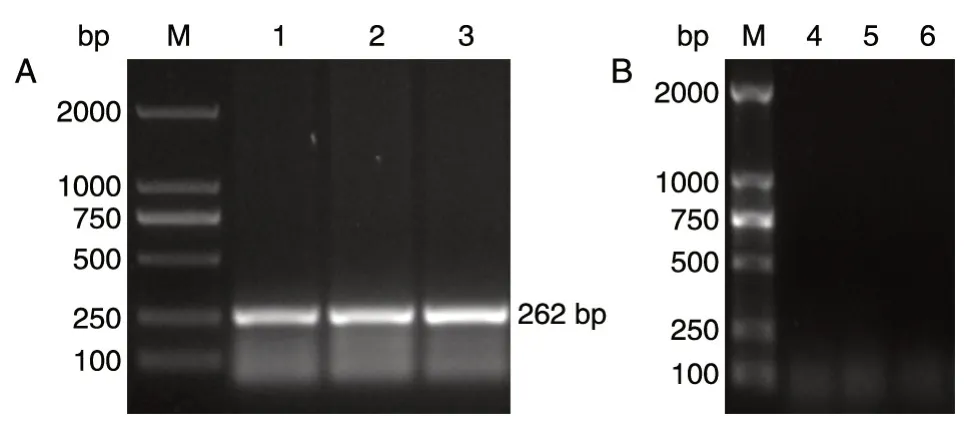
图6 琼脂糖凝胶电泳验证kanMX、pdc1、pdc5 基因

图7 工程菌株的发酵变化曲线
2.4 重组菌株摇瓶发酵试验
图7A 所示,菌株H5/H5-01/H5-02 发酵12 h后进入对数生长期,36 h 达到稳定期,但重组菌株H5-01 和H5-02 的长势略低于H5,D600nm值较原始菌株分别降低了16.89%、13.80%。出现这一结果的原因可能是敲除pdc1和pdc5后不同程度影响了菌株生长及体内的酸碱平衡,进而使菌株生长呈现缓慢趋势。在0~12 h 时pH 值总体呈上升趋势,但24 h 后重组菌株的pH 值高于原始菌株,可能是重组菌株的丙酮酸脱羧酶基因被破坏,导致其乙酸产量降低,使其pH 值略高。图7B 所示,经HPLC 检测发现重组菌株乙醇产量明显低于原始菌株,H5-01、H5-02 的最高乙醇产量分别为35.946±0.918、34.680±0.887 g/L,较原始菌株H5 乙醇产量(42.056±0.880 g/L)分别下降了14.53%与17.54%。此外,重组菌株的2,3-BD 积累量明显高于原始菌株,最高产量为0.264±0.015 g/L。由此可见,含有pdc1、pdc5同源序列线性片段的敲除质粒pTWCL-PDC1 与pTWCL-PDC5 构 建成功,导致重组菌株的丙酮酸脱羧酶活性降低,乙醇产量明显下降,并且流向乙醇的部分碳源流向了2,3-BD 的生成方向,使其2,3-BD 产量得到提高。
3 讨论
作为一种单细胞真核生物,酿酒酵母易于培养,其遗传操作技术成熟,相关理论研究较为清楚,可以利用基因工程手段对酿酒酵母进行更加准确的改造[15-16]。其中,利用Cre-loxP 重组酶技术可以准确、高效地敲除靶基因,而且标记基因也可以被选择性切除。林晓华等[17]扩增带有kanr筛选标记的SNF4基因敲除组件,转化酿酒酵母,利用Cre-LoxP 重组酶技术切除kanr筛选标记并获得SNF4等位基因完全缺失菌株。Semkiv 等[18]从大肠杆菌中克隆获得kanMX4,将该基因与报告基因PHO8(编码碱性磷酸酶)相连,构建多拷贝数的整合表达载体并转化酿酒酵母,使其具有更高的抗生素抗性。本研究通过PCR 克隆得到pdc1-loxP-kanMX基因片段,采用同源重组原理构建表达载体,该载体含有基因敲除组件与筛选标记基因(loxP-kanMX-loxP),在一定程度上可作为敲除酿酒酵母相关基因的技术参考。
丙酮酸是糖酵解过程中的关键中间产物,连接许多代谢途径,PDC 是酿酒酵母代谢途径中的关键调节酶,其活性主要由pdc1、pdc5和pdc6这3个基因控制,可以使丙酮酸生成乙醛,然后通过醛醇缩合反应合成少量的乙偶姻,并进一步生成2,3-BD[19-20]。我们构建了含有pdc1、pdc5同源序列线性片段的敲除质粒pTWCL-PDC1 与pTWCLPDC5,并将其分别转化酿酒酵母H5,通过摇瓶发酵试验发现重组菌株的乙醇产量较原始菌株明显下降,而2,3-BD 产量明显上升,表明构建后的质粒对酿酒酵母的PDC 活性产生了影响。丙酮酸在PDC 作用下生成乙醛,乙醛又能生成乙醇[21],质粒pTWCL-PDC1 与pTWCL-PDC5 使酿酒酵母H5 的pdc1、pdc5基因被分别敲除,菌株的丙酮酸脱羧酶活性减弱,中间产物丙酮酸大量积累,减少了乙醇的产生,使碳源更多流向2,3-BD 产生途径。吴满珍等[22]构建含有pdc1和adh1同源序列的质粒,将其导入酿酒酵母CEN.PK2-1C,重组菌株的最高乙醇产量较野生菌株下降了63.8%,而丙酮酸达到2.0 g/L。Lian 等[23]构建了质粒pRS426,通过同源重组方法使野生型酿酒酵母体内同时缺失pdc1、pdc5、pdc6基因,并过表达转录因子MTH1,利用葡萄糖和半乳糖作为底物,通过补料分批发酵,其2,3-BD 产量提高到100 g/L。
综上,我们依据NCBI 获得pdc1、pdc5基因左右两端各40 bp 同源序列,以pUG6 质粒DNA 为模板设计引物,PCR 分别扩增两端各含40 bppdc1、pdc5同源序列的基因敲除片段pdc1-loxP-kanMX、pdc5-loxP-kanMX,分别插入质粒pMD18-T、pEASY-T3,得到重组表达载体pTWCL-PDC1与pTWCL-PDC5,并将其分别转化酿酒酵母H5,摇瓶发酵试验发现重组菌株乙醇产量明显下降,PDC 活性降低。研究结果表明,含有酿酒酵母pdc1、pdc5同源序列的loxP-kanMX-loxP质粒构建成功,酿酒酵母中的pdc1、pdc5基因被成功敲除。
猜你喜欢
山西医科大学学报(2022年6期)2022-08-04
酿酒科技(2021年8期)2021-12-06
牡丹江医学院学报(2021年5期)2021-12-05
汉字汉语研究(2021年2期)2021-08-30
军事文摘·科学少年(2021年1期)2021-02-04
酿酒科技(2020年7期)2020-12-19
河南化工(2020年2期)2020-04-20
汉字汉语研究(2019年2期)2019-08-27
食品安全导刊(2018年30期)2019-01-28
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
