
一种检测慢病毒滴度的实时荧光定量PCR方法
2019-09-27 02:30张飞飞孙文耿琦丁怡彤
生物技术通讯 2019年4期
张飞飞,孙文,耿琦,丁怡彤
徐州医科大学 麻醉学院,江苏省麻醉学重点实验室,江苏 徐州221004
慢病毒载体是以人免疫缺陷Ⅰ型病毒(HIV-1)为基础发展起来的反转录病毒载体,与传统的病毒载体相比,对分裂细胞和非分裂细胞均具有感染能力[1-2],具有自我失活功能,是无免疫反应的高效基因传递工具,容纳的外源基因片段大、宿主范围广、重组机会低,可将所携带的目的基因整合到宿主染色体中并稳定长期表达[3-4],随亲本的繁殖将外源基因传递给子代,是目前应用最广泛的基因运载工具之一,已用于转基因动物的制备和基因治疗研究中。
通常用病毒滴度来衡量慢病毒制备是否成功以及慢病毒的质量,因此滴度测定是一个重要的步骤,尤其是用于基因治疗或转基因动物研制等需要准确测定滴度的重组慢病毒。目前测定慢病毒滴度的主要方法有流式细胞分选法(FACS)[5-6]和p24 蛋白酶联免疫法(ELISA)[7-8]。通过流式细胞仪检测靶细胞中被感染的细胞的比例,能准确反映病毒滴度以及病毒的实际感染活力,但所需设备昂贵,而且仅限于表达荧光报告基因的慢病毒载体。p24 蛋白ELISA 法检测的是病毒的物理单位,即不能区分具有感染能力和不具有感染能力的病毒颗粒,并不能反映病毒的真实感染活力,因此该方法的应用也受限制。鉴于此,建立一种特异、灵敏、有效的慢病毒滴度分子检测方法,可为慢病毒的应用提供技术支持。
在《分子克隆实验手册》提供的慢病毒载体测定方法基础上[9],本文介绍一种利用实时荧光定量PCR(RT-qPCR)测定慢病毒滴度的方法。该方法通过设计定量引物检测WPRE 序列(WPRE序列是重组慢病毒中常见的调控元件,且在正常情况下不存在于动物细胞体内,因此可以作为重组慢病毒插入靶细胞基因组后的一个定量检测标志物),同时选择人源性的单拷贝基因白蛋白(albumin,Alb)基因作为内参基因[10-11],最终计算得到每个细胞整合的慢病毒拷贝数。该方法检测到的是感染了细胞并成功整合的病毒,能体现出病毒的真实感染活力,且不受细胞基因组DNA提取效率影响,另外荧光标签的有无对滴度检测没有影响。
1 材料与方法
1.1 材料
HEK293T 细胞购自中国科学院上海生命科学研究院细胞库;大肠杆菌DH5α、Stbl3,克隆质粒pBluescript SK(+),包膜质粒pMD2.G,包装质粒psPAX2,转移质粒pLentiCRISPR V2 均为本实验室保存;限制性内切酶、T4DNA 连接酶、胶回收试剂盒、PCR 产物纯化试剂盒、DNA marker 及SYBR Green 荧光染料购自大连宝生物公司;无内毒素大提试剂盒和细胞基因组提取试剂盒购自天根生化科技有限公司;胎牛血清(FBS)、胰蛋白酶(0.25% Trypsin-EDTA)、LipofectAMINE 2000、OPTI-MEM 和DMEM 培养基购自Gibco 公司;PCR引物合成及测序由苏州金唯智公司完成;实时荧光定量PCR 仪为Roche 公司的LightCycler 480II;超速离心机为Beckman Coulter 公司的Optima L-100XP;核酸浓度测定仪为Thermo Scientific 公司的NanoDrop 2000;真空离心浓缩仪为Genevac 公司的miVac Duo;生物安全柜为Heal Force 公司的HFsafe-1200LCB2。
1.2 分子生物学常规操作
大肠杆菌的培养、PCR 扩增、质粒提取和酶切鉴定等实验参照文献[12]进行。本研究所用PCR引物序列见表1。
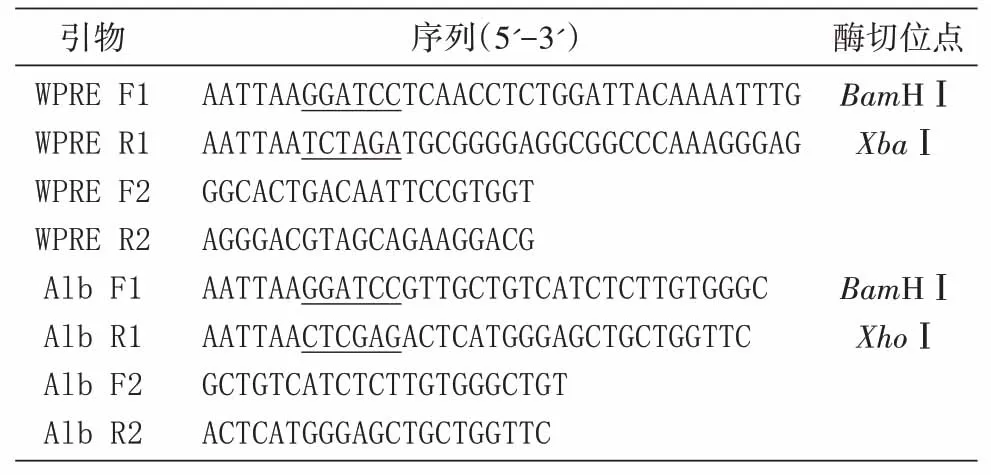
表1 本研究所用PCR引物
1.3 WPRE 元件和Alb 基因的质粒构建
以pLentiCRISPR V2 质粒为模板、WPRE F1和WPRE R1为引物,扩增出588 bp 的WPRE 序列,PCR 产物经BamHⅠ和XbaⅠ酶切后,克隆到pBluescript SK(+)的相同位点,经酶切和测序验证后命名为pSK-WPRE。以HEK293T 细胞基因组DNA 为模板、Alb F1 和Alb R1 为引物,扩增出142 bp 的Alb 基因序列,PCR产物经BamHⅠ和XhoⅠ酶切后克隆至pBluescript SK(+)/BamHⅠ-XhoⅠ获得pSK-Alb,酶切鉴定并测序。
1.4 计算标准品质粒的拷贝数
根据公式①计算标准品的拷贝数:
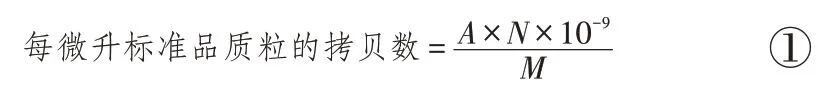
其中,A为标准品质粒的质量(ng),N为阿伏伽德罗常数6.02×1023,M为标准品的相对分子质量(标准品的碱基对数×1 对碱基的平均相对分子质量660)。
pSK-WPRE 质粒大小为3540 bp,浓度为500 ng/μL,计算pSK-WPRE 的拷贝数为1.29×1011拷贝/μL。同理,pSK-Alb(3055 bp,500 ng/μL)的拷贝数为1.49×1011拷贝/μL。最终稀释获得拷贝数分别为1.25×106、1.25×105、1.25×104、1.25×103拷贝/μL 的4 个梯度的标准品质粒。
1.5 慢病毒的包装
1.5.1 质粒转染和慢病毒收集 采用LipofectAMINE 2000 共转染293T 细胞,转染前24 h,将293T 细胞以5×106/10 cm 培养皿密度接种,加入10 mL 293T 培养基,在37℃、5% CO2条件下培养,转染前细胞密度应达到80%~90%。培养6 h后弃去含转染混合物的培养基,PBS 清洗一次,加入含10%血清的新鲜完全培养基,分别于24、48、72 h 收集富含慢病毒颗粒的细胞上清培养液。
按照如下反应体系配置:22.5 μg pLenti-CRISPR V2,16.9 μg psPAX2,5.6 μg pMD2.G,加 OPTI- MEM补至500 μL;125 μL LipofectAMINE 2000 转染试剂,375 μL OPTI-MEM,总体积500 μL。
1.5.2 慢病毒浓缩和纯化 将收集的293T 细胞上清液于4℃、4000 r/min 离心10 min,去除细胞碎片,用0.45 μm 的过滤器过滤,获得的上清液于4℃、82 700 r/min 超速离心2 h,弃上清,加入PBS,轻轻反复吹打重悬,充分溶解后分装。
1.6 慢病毒感染293T 细胞
检测前1 d 将293T 细胞传代,24 孔板中每孔加入2.5×104细胞,体积为500 μL;根据病毒的预期滴度准备3 个无菌EP 管,每管加入90 μL 培养基(10% FBS+DMEM);取待测定的病毒原液10 μL 加入第1 管,混匀后取10 μL 加入第2 管,继续相同操作到第3 管;选取所需细胞孔,移除培养基并用PBS 清洗,加入90 μL 病毒稀释液、410 μL 生长培养基和5 μg/μL Polybrene 进行培养;感染24 h 后移除含病毒的培养基,换为500 μL含DNaseⅠ的新鲜培养基,37℃消化15 min(这一步是要除去慢病毒悬浮液里残余的质粒DNA,防止影响后续慢病毒滴度测定),然后加入新鲜的生长培养基,小心操作,不要吹起细胞;2~3 d 后,抽提基因组DNA。
1.7 实时荧光定量PCR
反 应 体系包括SYBR Green master(2×)10 μL,上、下游引物(WPRE F2-WPRE R2 或Alb F2-Alb R2,10 μmol/L)各1 μL,基因组DNA 模板100 ng 或104~107拷贝的标准品8 μL,补水至20 μL。扩增程序:95℃预变性30 s,95℃变性5 s,60℃退 火30 s,循环40 次。熔 解 曲线 程 序:95℃ 5 s,60℃ 1 min,95℃,1 次循环,退火延伸时检测荧光信号。每个样品做2 个重复,同一个样品进行3 次独立的荧光定量PCR 检测。
1.8 慢病毒滴度计算方法
分别以慢病毒感染的细胞基因组和104~107拷贝的标准品为模板进行定量PCR,并根据标准品的Ct 值和拷贝数绘制标准曲线,将待测样品的Ct 值代入标准曲线,计算获得待测样品的WPRE和Alb 的拷贝数。根据公式②计算得到每个细胞整合的慢病毒拷贝数,最后根据公式③算得慢病毒滴度。

2 结果
2.1 标准品质粒的构建
重组质粒pSK-WPRE 和pSK-Alb 分别经BamHⅠ/XbaⅠ和BamHⅠ/XhoⅠ双酶切鉴定,结果见图1,均与预期大小一致,表明标准品质粒构建成功。
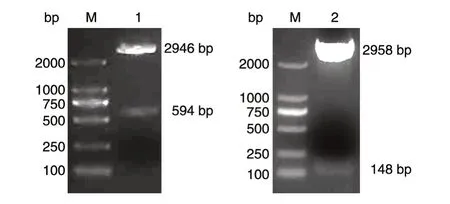
图1 标准品质粒酶切鉴定
2.2 标准曲线的建立
取4 个不同稀释比例(104~107拷贝)的标准品质粒作为模板进行荧光定量PCR,获得的扩增动力学曲线图如图2A。根据扩增曲线图,软件分别生成pSK-WPRE 和pSK-Alb 质粒的标准曲线(图2B),回归方程分别为y=-4.255x+46.047 和y=-2.8735x+35.831,相关系数R2均大于0.99,表明在标准品质粒4 个不同稀释比例范围内具有非常好的线性关系,扩增效率E均大于95%。最终扩增产物的熔解曲线如图2C,波峰单一,表明PCR 产物特异性较好,另外波峰的高度与标准品DNA 的拷贝数呈正相关。
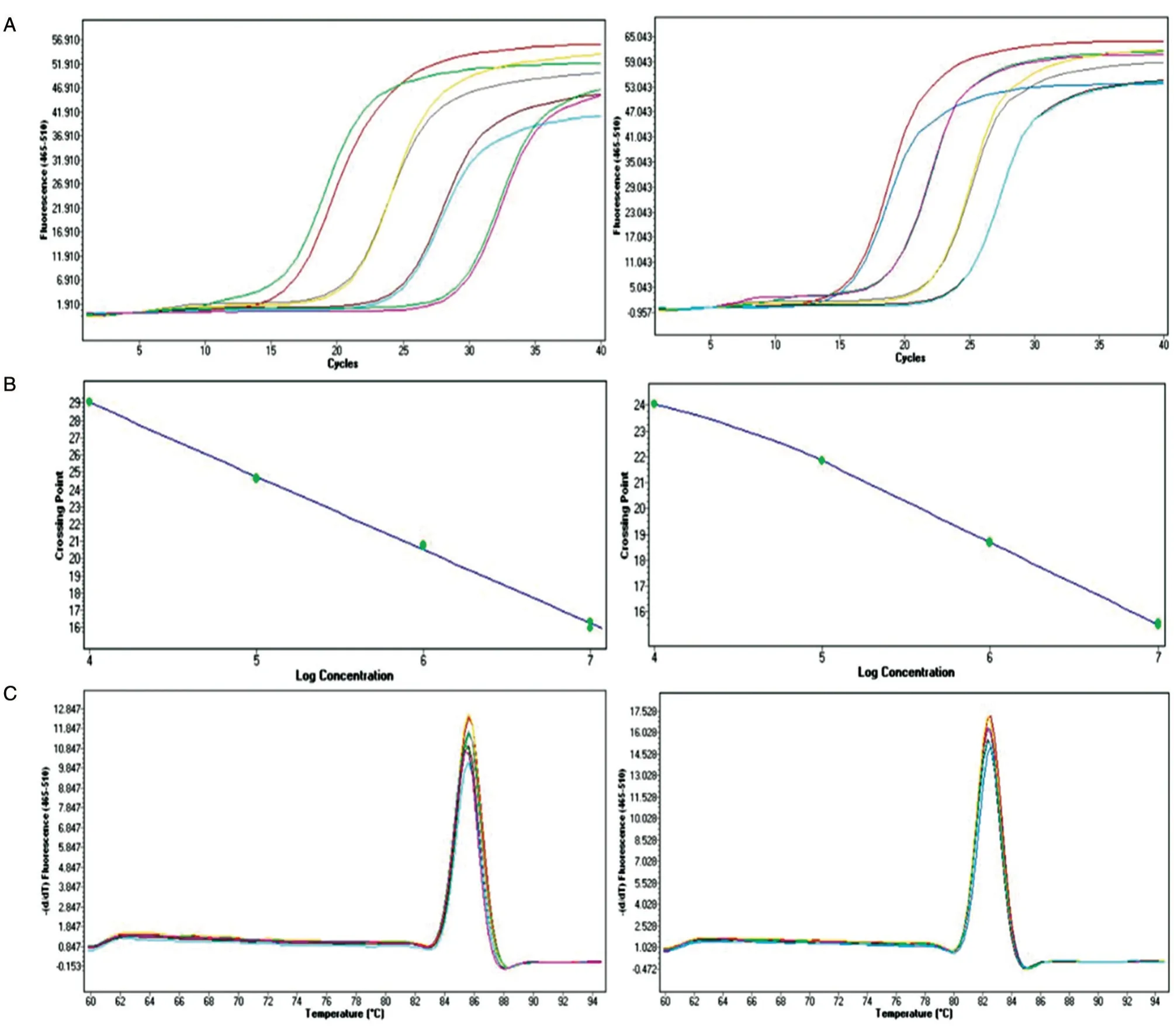
图2 实时荧光定量PCR 检测pSK-WPRE 和pSK-Alb 标准品质粒
2.3 实时荧光定量PCR 检测慢病毒整合拷贝数并计算慢病毒滴度
根据1.5 的操作流程包装慢病毒,获得的慢病毒按照1.6 所述方法稀释,分别获得体积为10、1、0.1 μL 的慢病毒稀释液,感染293T 细胞2 d后,提取细胞基因组DNA,应用RT-qPCR 检测不同稀释比例的慢病毒感染的细胞基因组DNA 中WPRE 整合拷贝数,结果见表2,最终的慢病毒滴度为(4.3±0.9)×106TU/mL。
3 讨论
慢病毒载体是近年出现的一种新的基因转导工具,由于其具有可以长期、稳定、高水平地表达,感染效率高,自身抗原性弱,可操控性强等优点,被应用于细胞和动物模型建立[13-14]以及基因治疗[15-16]等领域。

表2 定量PCR法测定慢病毒滴度
高滴度慢病毒的包装受很多因素的影响,经过多次的包装实验,本课题组总结获得高滴度慢病毒的关键在于:①保障包装细胞系HEK293T 的质量,传代次数尽可能少;②转染时合适的细胞融合率,细胞量过少不容易包装出病毒,过大则细胞会在病毒收集前因为接触抑制而大量死亡,也不容易包装出病毒,最佳细胞融合率为60%~70%;③不含内毒素的包装质粒浓度大于500 ng/μL,且D260nm/D280nm>1.8,D260nm/D230nm>1.8;④转 染 后24、48 和72 h 分3 次收集病毒上清,采用超速离心方法进行慢病毒浓缩。
高滴度的慢病毒是其实现高效基因传递的基础,所以准确测定慢病毒滴度尤为重要。已有文献报道利用RT-qPCR 方法检测慢病毒滴度。Sanburn 等[17]采用测定慢病毒悬浮液中的RNA 水平方法,但往往由于缺陷的病毒颗粒导致过高评估慢病毒滴度。Lizee 等[11]通过检测慢病毒侵染细胞的mRNA 水平,但有时慢病毒的功能原件很有可能整合到不适合基因转录的基因组区域,从而低估了慢病毒滴度。这2 种方法均涉及到RNA的提取及逆转录过程,存在操作复杂及重复性差等问题。本研究所阐述的基于SYBR Green 的实时荧光定量PCR,是以被病毒侵染的细胞基因组DNA 为模板,首先体外DNA 比RNA 更加稳定,其次基因组DNA 的提取更加方便快捷。
和基于荧光探针的定量PCR 测定慢病毒滴度的结果相类似[18],本研究基于SYBR Green 的定量PCR 方法的扩增效率接近100%,熔解曲线分析无引物二聚体。
最终我们只获得滴度为106数量级的慢病毒,和商品化方法获得108甚至109数量级的慢病毒还相差甚远。我们猜测pLentiCRISPR V2 慢病毒载体上5'长末端重复序列(LTR)和3'LTR 之间有将近10 kb 的病毒调控元件和Cas9 基因,如此长的片段导致其包装效率和感染力比一般慢病毒低。但总的来说,本方法成本低、操作简便快捷、重复性好、准确性高,为重组慢病毒应用于临床基因治疗和CRISPR-screening 等需要准确慢病毒滴度的相关研究奠定了良好的基础。
猜你喜欢
右江民族医学院学报(2022年2期)2022-05-19
昆明医科大学学报(2022年2期)2022-03-29
河北医学(2021年10期)2021-10-27
华侨大学学报(自然科学版)(2021年4期)2021-07-30
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国临床医学影像杂志(2019年6期)2019-08-27
中成药(2018年12期)2018-12-29
吉林医学(2018年12期)2018-12-21
中成药(2017年6期)2017-06-13
中华实验和临床病毒学杂志(2016年3期)2016-08-09
