
海洋环肽stylissamide Ⅰ的全合成研究
2019-09-25 02:06陆东涛秦路平
药学实践杂志 2019年5期
陆东涛,刘 超,秦路平,邹 燕
(海军军医大学:a.长海医院药材科,b.药学院,上海 200433)
海洋天然产物因有其独特的化学结构和良好的生物活性,许多课题组都致力于从其中寻找具有潜在药理活性的先导化合物,迄今为止,许多化合物已经成为药物化学研究的热点[1-2]。在海洋天然产物中,环肽是有着独特药理活性和生物化学特性的一类化合物[3-4]。这类化合物因其特殊的稳定性和选择性,能够有效地模仿蛋白表位来干扰蛋白-蛋白间相互作用,体现出较高的临床治疗价值[5]。与传统直链肽相比,环肽类化合物具有结构稳定、脂溶性高、穿膜性强、体内半衰期长等优点[6-8],使其在抗结核[9]、抗菌[10]、抗病毒[11-12]、抗肿瘤[13]、抗炎[14-15]等药物的研发中发挥着无可替代的重要作用。
Stylissamide是从Stylissa属海绵中分离得到的一类富含脯氨酸残基的环肽化合物,自2007年Schmidt等[16]从Stylissacaribica中分离得到stylissamide A以来,已经先后从该属海绵中得到一系列类似环肽化合物,该系列环肽表现出抗肿瘤和抗菌等多种生理活性[17-18]。Stylissamide Ⅰ是由Kubota等[19]从日本冲绳海绵Stylissasp.样品中提取纯化得到的环七肽化合物,经过高分辨电喷雾质谱以及二维核磁等技术表征,明确其序列为Cyclo-(Pro-Pro-Val-Pro-Tyr-Tyr-Tyr),结构如图1所示。进一步研究发现,stylissamide Ⅰ对Aspergillusniger显示出较好的抑制活性,是具有开发前景的抗真菌药物先导化合物。然而,单纯依靠从天然产物中分离纯化得到stylissamide Ⅰ很难满足对其药理活性和构效关系等进一步研究的需要,并且也无法对其开展结构优化和修饰等研究。因此,一种高效简便的全合成方法是十分必要的。
以往对类似环肽化合物的固相合成/液相环合研究中,通常使用1-羟基苯并三唑(HOBt)/N,N-二异丙基碳二亚胺(DIC)作为缩合剂完成直链多肽的固相合成[20-21]。该方法氨基酸偶联形成酰胺键所需反应时间较长(2~4 h),并且在缩合过程中DIC所形成的副产物N,N-二异丙基脲溶解性差,需要使用大量有机试剂冲洗树脂,后处理烦琐。本研究基于课题组前期研究成果,以二氯树脂为固相载体,通过缩合剂DIPEA/HCTU依次连接Fmoc/tBu保护的氨基酸,完成多肽的固相合成[22];随后在三氟乙醇的条件下将直链肽从树脂上切割下来,后通过环合试剂PyAOP/HOAt/NMM在溶液中完成环合;最后利用三氟乙酸脱去侧链保护基获得环肽粗品。经反相高效液相色谱对所合成的环肽粗品进行纯化,首次成功完成对stylissamide Ⅰ的全合成,合成路线如图2所示。

图1 stylissamides I的结构

图2 stylissamides Ⅰ的合成路线
合成试剂与条件:a.Fmoc-Pro-OH,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,120 min; b.(Ⅰ) 20% 哌啶/N,N-二异丙基乙胺,30℃,10 min,重复2次;(Ⅱ) Fmoc-Tyr(OtBu)-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(Ⅲ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次;(Ⅳ) Fmoc-Tyr(OtBu)-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(Ⅴ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次;(Ⅵ) Fmoc-Tyr(OtBu)-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(Ⅶ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次;(Ⅷ) Fmoc-Pro-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(Ⅸ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次;(Ⅹ) Fmoc-Pro-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(Ⅺ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次;(Ⅻ) Fmoc-Val-OH,6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,30℃,40 min;(ⅩⅢ) 20% 哌啶/N,N-二甲基甲酰胺,30℃,10 min,重复2次; c.三氟乙醇/二氯甲烷(1∶4,V/V),30℃,4 h; d.六氟磷酸(7-氮杂苯并三唑-1-氧基)三吡咯烷磷,N-羟基-7-氮杂苯并三氮唑,N-甲基吗啡啉,N,N-二甲基甲酰胺,二氯甲烷,室温,12 h; e.三氟乙酸/二氯甲烷(1∶3,V/V),室温,2 h。
1 材料与方法
1.1 仪器
LOOYE ZX98-1型旋转蒸发仪、2ZX-4型旋片真空泵、低温冷却循环泵、SHB-ⅢA型循环水式多用真空泵(上海豫康科教仪器设备有限公司),CHA-S型气浴恒温振荡器(江苏金坛市国胜实验仪器厂),高效液相色谱系统及紫外检测器(日本岛津公司),冷冻干燥机(美国LABCONCO公司),6538 UHD Accurate-Mass Q-TOF LC/MS(美国Agilent公司),Bruker DRX 600 MHz型高分辨核磁共振谱仪(德国Bruker公司)。
1.2 主要试剂
Fmoc-氨基酸、二氯树脂(吉尔生化上海有限公司); PyAOP,HCTU,HOAt,NMM(Adamas试剂公司); DIPEA(Acros试剂公司); TFE,TFA(百灵威科技有限公司);N,N-二甲基甲酰胺(DMF)、二氯甲烷(DCM)、甲醇、哌啶(国药集团化学试剂有限公司); 乙腈(J.T.Baker试剂公司);双蒸水(自制)。
1.3 方法
1.3.1树脂的活化
将二氯树脂1.0 g(载量0.3 mmol/g)加入到固相反应器中,加入DCM与DMF各5 ml浸泡10 min后抽滤,使树脂充分膨胀。
1.3.2Fmoc-氨基酸与树脂的连接
将 Fmoc-Pro-OH(337.0 mg,1.0 mmol,3.3 倍当量),DIPEA(387.6 mg,3.0 mmol,10.0倍当量) 的DMF溶液加入到固相反应器中,于气浴恒温振荡器中30℃反应120 min。反应结束后,用DCM与DMF洗涤树脂各5遍。
1.3.3Fmoc的脱除
每一步生成肽键反应之前,均需脱除Fmoc保护。向固相反应器中加入6 ml 20 %的哌啶/DMF溶液,于气浴恒温振荡器中30℃反应10 min,抽滤后再次加入6 ml 20 %的哌啶/DMF溶液反应10 min。反应结束后,用DCM与DMF洗涤树脂各5遍。
1.3.4肽键的生成
所有后续Fmoc保护的氨基酸采用活化酯方法缩合。向连接在树脂上脱去末端Fmoc保护的直链肽中加入Fmoc-氨基酸(1.0 mmol,3.3倍当量),HCTU(413.7 mg,1.0 mmol,3倍当量),DIPEA(387.6 mg,3.0 mmol,10.0倍当量)的DMF溶液,于气浴恒温振荡器中30℃反应40 min,反应完毕后,用DCM与DMF洗涤树脂各5遍。重复此过程以及脱保护过程直至所有氨基酸全部接入。
1.3.5多肽的切割
向固相反应器中加入TFE/DCM(1∶4,V/V) 混合溶液10 ml,于气浴恒温反应器中30℃反应4 h。抽滤收集滤液,于旋转蒸发仪蒸干溶剂,乙醚打浆后真空泵干燥,获得直链肽粗品273.1 mg。
1.3.6直链肽的环合与凝胶柱纯化
将直链肽粗品溶于DCM溶液(290.0 ml),于0℃缓慢滴加至含有PyAOP(783.0 mg,1.5 mmol,5.0倍当量),HOAt(204.2 mg,1.5 mmol,5倍当量),NMM(303.5 mg,3.0 mmol,10倍当量)的DMF溶液(10 ml)中。滴加完毕后于室温反应12 h,反应结束后蒸干溶剂,得到淡黄色油状液体。将其用甲醇溶解后,采用葡聚糖凝胶LH-20柱对其进行初步纯化,获得带侧链保护基的环肽粗品243.4 mg。
1.3.7侧链保护基的脱除和纯化
将带侧链保护基的环肽粗品溶解于TFA/DCM(1∶3,V/V) 的混合溶液中,于室温反应2 h后旋干溶剂。采用反相高效液相色谱对其进行纯化。色谱条件:SHIMADZU PRC-ODS柱(50 mm×250 mm,15 μm ) ;流动相A:乙腈+0.1%TFA,流动相B:水+0.1%TFA,线性洗脱梯度 B :100%~10%,洗脱时间30 min,流速6.0 ml/min;紫外检测器检测波长214 nm。获得纯品176.7 mg,总收率67%。
2 结果与讨论
2.1 stylissamide Ⅰ的HPLC表征
本研究以水和乙腈为流动相进行梯度洗脱,对stylissamide Ⅰ粗品进行纯化。粗品HPLC图中峰数较多,主峰物质含量为63.5%。通过制备色谱对主峰进行纯化,色谱图中主峰明显,含量为98.9%(图3)。
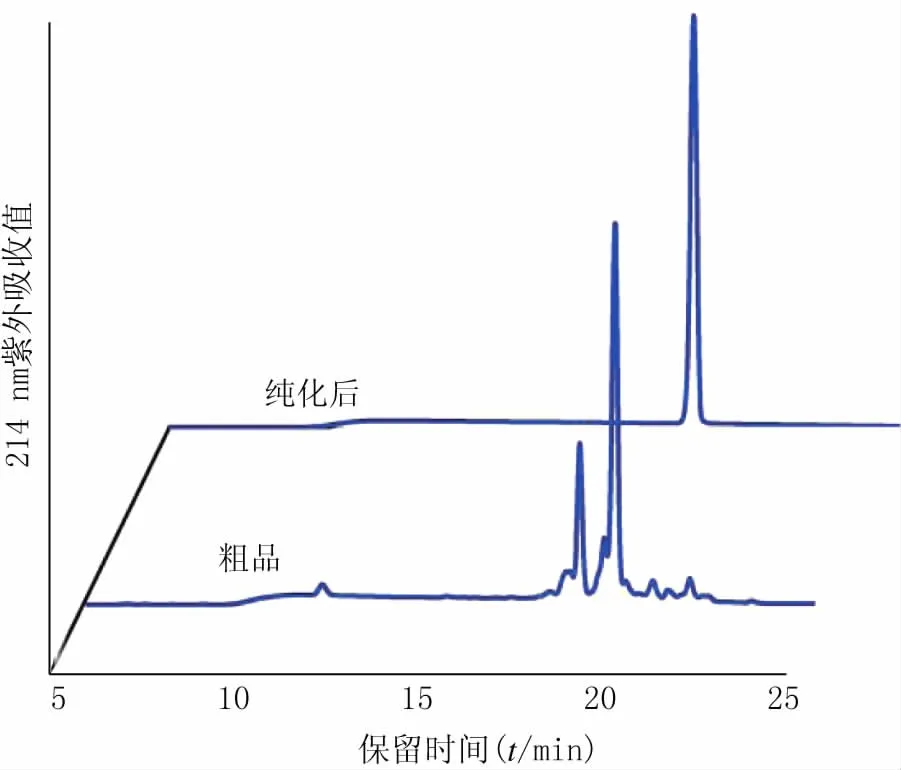
图3 stylissamide Ⅰ粗品与纯品的HPLC图谱
2.2 stylissamide Ⅰ的高分辨质谱表征
通过Q-TOF LC/MS分析,质谱图中880.426 4对应[M+H]+峰,902.402 4对应[M+Na]+峰(图4),与stylissamide Ⅰ理论分子量879.416 7相比,误差值为1.1×10-5,显示主峰物质分子量与stylissamide Ⅰ相符。随后,采用MassHunter Workstation(Agilent Tech)质谱工作站对分子式进行匹配,分子式为C47H57N7O10,与理论分子式相符。
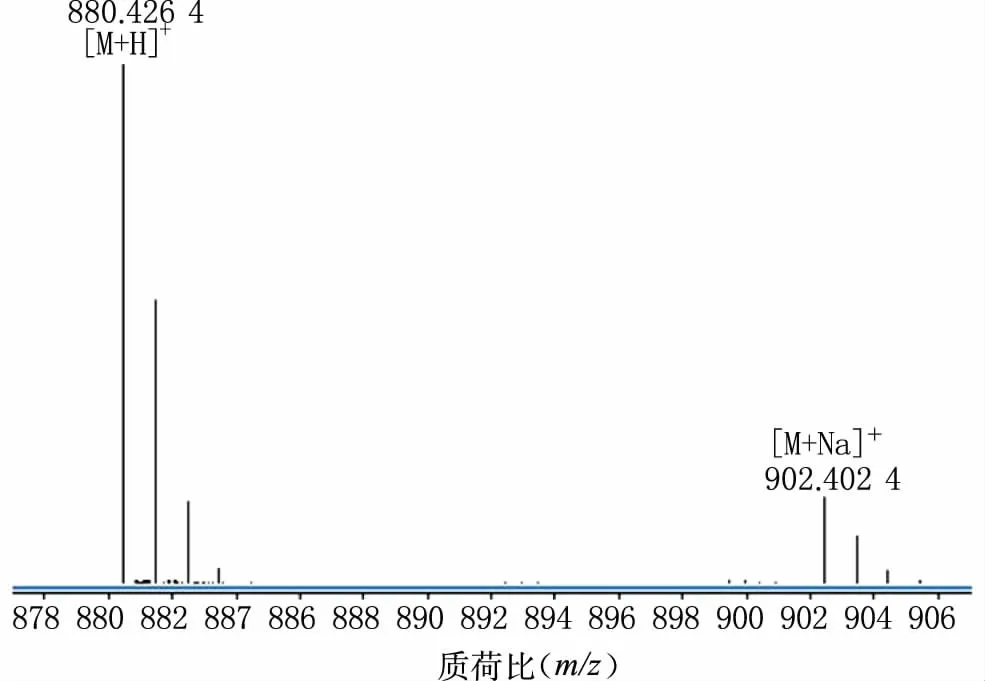
图4 stylissamide Ⅰ纯化后的高分辨质谱图
2.3 stylissamide Ⅰ的核磁共振氢谱与碳谱表征
本研究通过600 MHz核磁共振氢谱与碳谱对所合成环肽进行进一步表征。
1H-NMR(600 MHz,d-DMSO):δ 9.01(d,J=6.0 Hz,1H),8.61(d,J= 6.0 Hz,1H),8.28(d,J=6.0 Hz,1H),7.92(s,3H),7.81(d,J=6.0 Hz,1H),7.07-7.00(m,6H),6.71-6.62(m,6H),4.22-4.20(m,1H),3.89(s,1H),3.56-3.36(m,11H),3.01-2.98(m,1H),2.89-2.84(m,2H),2.73-2.65(m,3H),2.17-2.07(m,2H),1.99-1.80(m,11H),0.92-0.78(m,6H)。
13C-NMR(600 MHz,d-DMSO):δ 173.7,171.7,170.6,170.5,170.1,169.3,168.4,158.5,158.3,157.0,156.4,156.2,131.1,131.0,130.6,130.6,130.5,128.0,127.7,125.1,118.5,116.5,115.8,115.6,115.4,115.3,59.5,58.9,57.9,55.7,54.8,53.9,52.6,47.1,37.3,36.7,36.5,30.7,29.1,29.0,28.5,25.0,24.9,24.9,19.5,18.4,18.2。
2.4 环合反应的策略
2.4.1环合方式的选择
目前公认的多肽环合策略可分为液相环合与固相环合两种。液相环合策略在氨基酸连接完毕后,在弱酸的条件下将带有保护基的多肽切下,在溶液中进行环合后利用强酸切除侧链保护基团,需要进行反复的纯化并且可能产生二聚体或多聚体等副产物,使得其合成效率和合成总收率都较低。固相环合策略则是在树脂上完成直链多肽连接后,直接利用环合试剂在固相载体上进行环合,经过强酸切割后直接得到目标产物,无需反复的纯化步骤,同时树脂的假稀释作用避免了二聚体与多聚体的生成,是目前公认的优于液相环合的一种环合策略[23]。然而,固相环合条件较为苛刻,需要氨基酸残基侧链提供额外的羧基用于偶联树脂(如天冬氨酸或谷氨酸)。本研究中的stylissamide Ⅰ并不符合固相环合的条件,因此,课题组选择液相环合策略对其进行全合成。
2.4.2环合位点的选择
在环肽的合成中,环合位点的确定决定着合成的成败。环合位点选择不当会导致关环速率减慢,产生二聚化、低聚化以及差向异构化等副产物。一般来说,环合位点的确定应遵循4个原则[24]:①在通常情况下,应避免在N-烷基、α,α-二取代、β-取代等空间位阻大的氨基酸,如缬氨酸和异亮氨酸之间环合。然而,脯氨酸是一个特例,选择脯氨酸为关环位点也可以获得较好的环合效果。②应尽量选择在L构型和D构型的氨基酸之间环合,不同构型的氨基酸之间发生的环合反应速率比相同构型氨基酸之间更快。③可借助X-晶体衍射或分子模型来确定能促进环合反应的分子内氢键的位置,并选择此处为环合位点。④能引起分子骨架扭转的结构有利于环合,例如甘氨酸。综上所述,考虑到环合未知空间位阻对环合速率及产率的影响,本研究选择在脯氨酸之间进行环合。
2.4.3环合条件的确定
较为常用的环合试剂主要有HOAt、HOBt、PyAOP、PyBOP等。本研究选择PyAOP/HOAt为环合试剂对,选用NMM提供碱性环境,室温反应12 h,成功完成环合反应[25]。
液相环合策略的一个主要问题是环合过程容易形成分子间的二聚体和多聚体副产物,影响合成产率。我们通过高度稀释的方法,将直链肽溶液浓度控制在约1 mg/ml,并在低温条件下向环合试剂溶液中缓慢滴加,明显减少了线性或环状二聚体和多聚体的产生[26]。
3 结论
本研究使用二氯树脂为载体固相合成了直链七肽,而后将其从树脂上切下,利用环合试剂在溶液中完成环合,最后在酸性条件下脱除全部侧链保护基团,成功获得环肽粗品。经反相高效液相色谱纯化得到目标环肽stylissamide Ⅰ,产物纯度达到98.9%,合成总收率为67%。通过高分辨质谱、核磁共振氢谱与碳谱分析,确定合成产物为目标环肽stylissamide Ⅰ。本研究首次完成了环肽stylissamide Ⅰ的化学全合成,合成方法具有快捷、简便、高效的特点,为该环肽及其他类似环肽类化合物的固相全合成提供了参考。
猜你喜欢
食品研究与开发(2022年9期)2022-05-17
能源化工(2021年3期)2021-12-31
吉林农业(2018年18期)2018-09-25
中外医疗(2018年7期)2018-06-20
大陆桥视野·下(2017年2期)2017-03-30
山东工业技术(2016年16期)2016-08-22
医学美学美容·中旬刊(2015年2期)2015-10-21
中国塑料(2015年12期)2015-10-16
有机氟工业(2015年3期)2015-03-03
中国药房(2011年23期)2011-11-17
