
增容作用对聚乳酸/高密度聚乙烯合金发泡行为的影响
2019-09-25 06:50:18殷德贤周洪福王向东叶志殷
中国塑料 2019年9期
李 杨,殷德贤,朱 敏,周洪福*,王向东,叶志殷
(1.北京工商大学材料与机械工程学院,北京 100048;2. 塑料卫生与安全质量评价技术北京市重点实验室,北京 100048)
0 前言
PLA作为一种典型的可生物降解脂肪族聚酯,具有原料来源广、生物降解性好、机械强度高[1-2]等优点。但是PLA的结晶度和结晶速率较低;PLA的熔体强度和黏弹性较差;PLA的韧性较差,加工窗口窄[3-4]。因此,需要对PLA进行共混、扩链、填充等改性,克服上述的一些缺点,制备具有优异发泡性能的PLA发泡材料[5]。由于PE-HD具有良好的规整性、良好的结晶性能和力学性能,在PLA中加入PE-HD可以同时提高聚乳酸的结晶性能和冲击强度。此外,在PLA/PE-HD共混物中采用PE-HD作为分散相,可以在PE-HD和PLA之间提供丰富的界面,有可能成为多相胞孔成核点,从而大大改善PLA泡沫的发泡性能[6]278。据我们所知,目前还没有关于PLA/PE-HD共混物发泡行为的研究报道在我们的前期工作中,采用PE-HD来改善PLA流变性能,增强PLA的可发性,提高PLA的结晶能力,调控泡孔形态[6]278。
本文将采用GEMA作为PLA/PE-HD合金的相容剂,调控两相微观形貌,改善合金的结晶和熔体黏弹性,提高其发泡性能。
1 实验部分
1.1 主要原料
PLA,2003D,D - 异构体含量为4.3 %,密度为 1.24 g/cm3,熔体流动速率为 3.2 g/10 min(190 ℃,2.16 kg),美国 Nature Works公司;
PE-HD,5420 GA,熔体流动速率为 2.3 g/10 min (190 ℃, 21.6 kg),密度为 0.95 g/cm3,熔点(Tm)为 139.6 ℃,独山子石化有限公司;
GEMA,熔体流动速率为5 g/10 min(190 ℃,2.16 kg),密度为0.94 g/cm3,甲基丙烯酸缩水甘油酯含量为8 %,Tm为104 ℃,法国Arkema公司。
1.2 主要设备及仪器
旋转流变仪,ARES Rheometer,美国TA仪器公司;
平板压片机,LP-S-50,美国Labtech公司;
电热鼓风干燥箱,DHG,上海一恒科学仪器有限公司;
密度天平,CPA2245,赛多利斯科学仪器(北京)有限公司;
傅里叶变换红外光谱仪(FTIR),NicoletIZ10,美国赛默飞世尔科技公司;
转矩流变仪,XSS-300,上海科创橡塑机械设备有限公司;
差示扫描量热仪(DSC), Q100,美国TA公司;
热台偏光显微镜(PLM),BX-51,日本Olympus公司;
扫描电子显微镜(SEM),Quanta FEG250,美国FEI公司;
高压发泡釜装置,200 mL,自制。
1.3 样品制备
将PLA、PE-HD和GEMA在80 ℃鼓风干燥烘箱中干燥4 h以去除水分;按表1将不同配比的PLA、PE-HD和GEMA在转矩流变仪中熔融共混,密炼温度为190 ℃,时间为15 min,转速为80 r/min;随后,在190 ℃和10 MPa压片机上模压成型10 min,制成厚度约1 mm的片材,冷却到室温获得PLA片材样品,用于进一步的表征和发泡过程。
表1 样品配方表
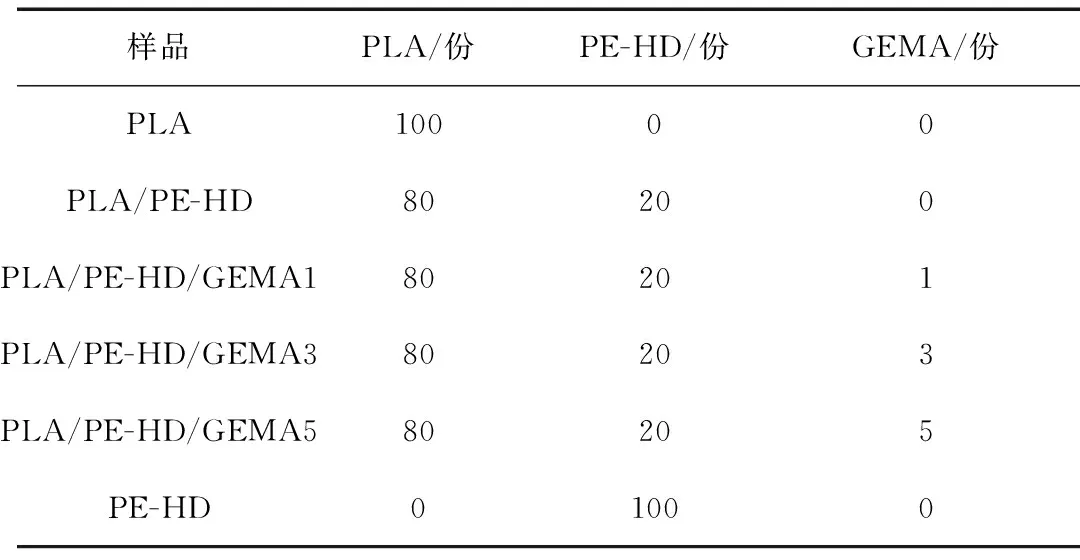
Tab.1 Formula of various samples
以CO2为发泡剂,采用间歇发泡法制备了PLA发泡样品,具体步骤如下:首先,将CO2注入高压釜1 min排除空气,待加热釜体到发泡温度(170 ℃),将PLA发泡样品置于高压釜中,然后将CO2注入高压釜内,压力达到20 MPa,浸泡2 h,待CO2在样品内溶解达到饱和,将温度分别冷却至95 ℃;然后在6 s左右释放CO2,高压釜的压力从20 MPa降至0.1 MPa,快速取出发泡样品并在室温下冷却定型。
1.4 性能测试与结构表征
DSC分析:采用DSC 对不同的PLA样品的结晶和熔融行为进行了研究,在N2气氛下(流速50 mL/min),取5~10 mg 的样品快速升温至190 ℃,保温5 min,消除热历史;以10 ℃/min 的降温速率降温至 40 ℃,保温5 min,再以10 ℃/min 的升温速率升温至190 ℃,得到样品的熔融与结晶曲线,并用式(1)和(2)计算各组PLA和PE-HD样品的结晶度(χc):
(1)
(2)
式中 ΔHm(PE-HD )——PE-HD的熔融焓,J/g
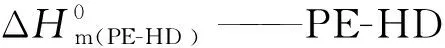
PLM分析:取少量试样放置于玻璃片间,以30 ℃/min的升温速率,将试样从室温升到200 ℃,保温5 min,尽量压薄,再以30 ℃/min的降温速率降至120 ℃,恒温40 min,观察试样的结晶情况;
FTIR分析:对不同的PLA样品进行红外光谱分析,每个光谱在3 800~480 cm-1范围内,波长分辨率为4 cm-1;
SEM分析:在液氮中浸泡4 h后,将PLA样品脆断,用SEM观察在10 kV加速电压下,不同PLA样品及其泡沫的断口形貌;观察前,样品表面喷涂一层金,以防止观察过程中电荷的积聚;
流变性能测试:样品的动态流变特性用旋转流变仪来表征,N2保护环境下,在直径为20 mm的圆形平行板间放置样品,平板测试间距为1 mm,最大应变在5 %,频率范围为 0.1~100 rad/s,测得在不同频率下的复数黏度(η*)、储能模量(G′)及损耗因子(tanδ);
发泡性能测试:PLA、PLA/PE-HD和PLA/PE-HD/GEMA发泡样品的发泡倍率(Φ)由式(3)计算:
(3)
式中ρf、ρp——样品发泡前和发泡后的密度,g/cm3
泡孔密度N0(个/cm3)由泡孔统计软件及式(4)统计计算可得:
(4)
式中n——SEM照片中泡孔个数
M——放大倍率
A——SEM照片中视野范围内的图片面积,cm2
2 结果与讨论
2.1 FTIR分析
图1是纯PLA、GEMA、PLA/PE-HD合金和PLA/PE-HD/GEMA 1合金的FTIR谱图。从图中可以看出,GEMA在2 918、1 463、720 cm-1处有3个环氧基团的特征峰。另外,PLA在1 750 cm-1处有很强的峰,这是其酯基基团上对应的峰。在加入GEMA之后PLA/PE-HD/GEMA1合金在2 918 cm-1处的峰值减弱,720 cm-1处的环氧特征峰值消失,说明GEMA上的环氧基团和PLA的基团(端羧基和羟基)发生了反应[7]。
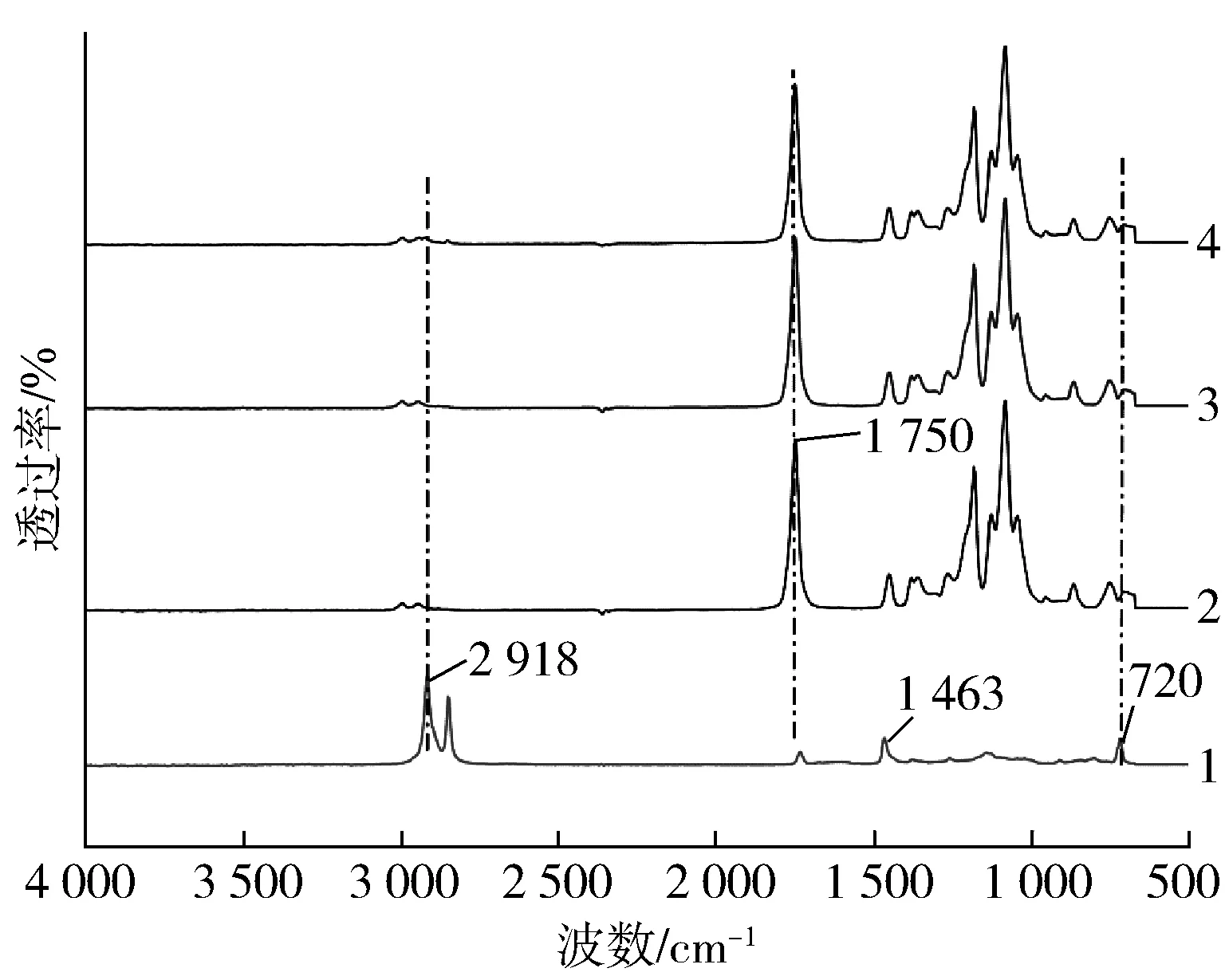
1—GEMA 2—PLA 3—PLA/PE-HD 4—PLA/PE-HD/GEMA1图1 PLA、GEMA、PLA/PE-HD和PLA/PE-HD/GEMA 1的FTIR谱图Fig.1 FTIR spectra of PLA,GEMA,PLA/PE-HD and PLA/PE-HD/GEMA1
2.2 DSC分析
图2是各样品在冷却和升温过程中的DSC曲线。由图2(a)可知,PLA降温曲线中没有明显的结晶峰,这是因为PLA分子链运动困难,在较快的冷却速率下结晶速率较慢导致的。相反,PE-HD在降温过程有明显的结晶峰,这应归因于PE-HD较高的结晶能力[8]。
在图2(b)中,PLA和PE-HD在(148.0±0.2) ℃和(132.1±0.1) ℃左右均有一个熔融峰。同时,在PLA的DSC升温曲线上出现了明显的冷结晶峰,这表明其结晶速率较慢[6]279。由表2可知,加入PE-HD后,PLA冷结晶峰消失,这可能是由于在PLA结晶之前,PE-HD结晶形成的微小晶体可以促进PLA的结晶,并且PLA/PE-HD合金的Tm略向高温移动。加入GEMA后,PE-HD的Tm变化不大,χc略微的增加。此外,PE-HD的加入, 使PLA的χc由(2.5±0.3)%提高到(9.3±0.2)%,但随着GEMA含量的增加,PLA的χc下降到(4.6±0.1)%。这可能是两方面的原因造成的;由于GEMA上的环氧基团和PLA的基团发生扩链反应,支化结构形成,分子链运动变的困难;另外,PE-HD分子链和GEMA的分子链缠绕在一起,造成了χc的下降。

1—PLA 2—PLA/PE-HD 3—PLA/PE-HD/GEMA1 4—PLA/PE-HD/GEMA3 5—PLA/PE-HD/GEMA5 6—PE-HD(a)降温曲线 (b)升温曲线图2 不同样品在10 ℃/min变温速率时的DSC曲线Fig.2 DSC curves of various samples at a cooling/heating rate of 10 ℃/min
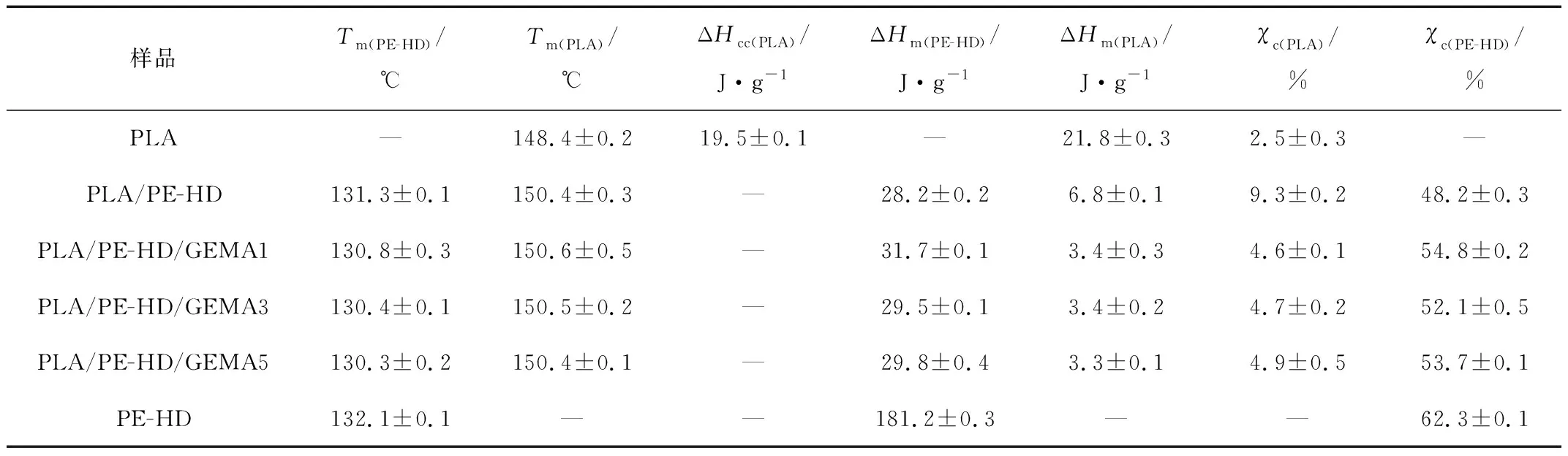
Tab.2 Thermal properties of various samples
图3是各样品在热台偏光显微镜下的的结晶形貌图,从图中可以看出,纯PLA的球晶数量较小,尺寸较大,球晶生长完整,而纯PE-HD由于结晶度较高而显示出大量的小碎晶。未加入GEMA之前,PLA/PE-HD的球晶数量较多,球晶尺寸较大。但是随着GEMA的含量增加球晶数量越来越多,尺寸越来越小。这是因为GEMA与PLA发生开环反应,生成大量的支化结构,这些支化点充当了异相成核点,使得球晶数量增加,但是由于支化结构的生成,使得分子链缠结,降低了分子链的运动能力,从而使得结晶不完全,球晶尺寸下降。大量的小球晶对于发泡会产生积极地作用,这些晶区与无定形区的界面能够充当泡孔成核点,提升泡孔密度;而大量小球晶的存在则能够提升试样的熔体强度,限制泡孔增长和合并。

(a)PLA (b)PLA/PE-HD (c)PLA/PE-HD/GEMA1 (d)PLA/PE-HD/GEMA3 (e)PLA/PE-HD/GEMA5 (f)PE-HD图3 不同样品的PLM照片Fig.3 PLM of various samples

(a)PLA (b)PLA/PE-HD (c)PLA/PE-HD/GEMA1 (d)PLA/PE-HD/GEMA3 (e)PLA/PE-HD/GEMA5图4 不同PLA样品的断面相貌形态Fig.4 Cryo-fracture surfaces of various PLA samples
2.3 分散相形态
图4是各样品的断面相貌形态图,在PLA基体中加入PE-HD后,由于PLA和PE-HD的相容性不好,PE-HD分散相在PLA基体中分散的不太均匀且粒径较大,可以清楚地观察到它们之间界面。在加入GEMA后,PE-HD分散相分散的更加均匀且粒径变小。随着GEMA含量的增加,分散相粒径逐渐变小,数量逐渐增加,分布的更加均匀,表明PE-HD与PLA的相容性增强。这可能是由于,GEMA上的环氧基团和PLA的基团发生扩链反应,支化结构的形成增加了两者界面结合力[6]280,[9],另外,分散相粒径变小,数量增加,对发泡材料的性能产生积极的效果,为发泡过程中泡孔的成核提供了异相成核点,进而影响泡孔尺寸、泡孔密度、发泡倍率。
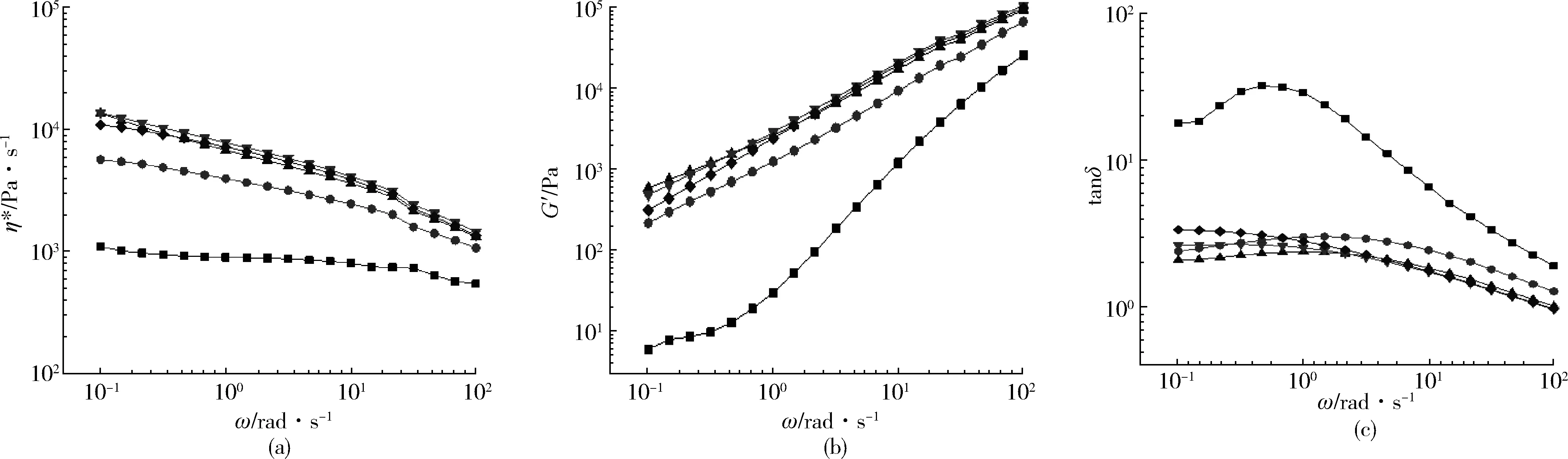
■—PLA ●—PLA/PE-HD ▲—PLA/PE-HD/GEMA1 ▼—PLA/PE-HD/GEMA3 ◆—PLA/PE-HD/GEMA5(a)η* (b) G′ (c) tan δ图5 不同PLA样品的动态流变特性Fig.5 Dynamic rheological properties of various PLA samples
2.4 流变性能分析
聚合物的发泡性能受聚合物的流变性能影响很大。图4是不同PLA样品的η*、G′、tanδ的变化曲线。在图5(a)中,在低频区PLA的η*较低,加入PE-HD之后,PLA/PE-HD合金的η*大幅度提高,这可能是由于PE-HD中分子链和PLA中线形分子链的缠绕所致。另外,PLA/PE-HD合金中η*随着GEMA的加入,进一步提高,这可能是由于支化结构的形成和界面结合力的增强所致[10]。
聚合物的可发性和聚合物熔体强度有关,而G′是表征熔体强度的重要指标之一。图5(b)为G′随角频率(ω)变化的曲线,在低频区,PLA/PE-HD合金的G′值高于PLA,说明加入PE-HD后,PLA熔体强度和弹性变好,PLA/PE-HD合金的可发性增强。加入GEMA后,体系的G′进一步提高,这是由于GEMA 的环氧基团在熔融共混过程中与PLA 的端基发生开环反应,形成支链结构使得分子链运动变得困难,同时又因为GEMA和PE-HD天然的相亲性,使得PLA相与PE-HD相容性增加,界面结合力增强,松弛时间较长,使体系的G′增加,更加适合于发泡[11]。
tanδ是在交变应力的作用下,熔体的应变滞后于应力,所受到的应力的相位差。从聚合物发泡角度来讲,在低频区,如果tanδ值越低,相位差就越小,那么体系的弹性响应越快,黏性耗散越不明显。从图5(c)可以看出tanδ随ω增加而不断减小,加入PE-HD和GEMA之后,在低频区tanδ明显下降,表明体系的弹性响应加快了,黏性耗散变小了,可发性提高。随着频率的不断提高,这种差别逐渐缩小。
2.5 发泡性能分析
由图6和表3可知,PLA发泡样品的泡孔尺寸较大,泡孔密度较小。 此外, PLA发泡样品的发泡倍率和泡孔尺寸分布也是最大的。这主要是由于PLA的结晶度小、熔体强度低造成的,泡孔成核少,泡孔增长快并出现泡孔合并现象。加入PE-HD之后,出现了复合泡孔结构,其中大泡孔和小泡孔的尺寸为50.0 μm和19.1 μm,对应的泡孔密度达到了9.9×106个/cm3,1.1×108个/cm3。出现这种情况的原因可能是,加入PE-HD之后,PLA的结晶度大幅度增加,而且PLA 与 PE-HD 两相界面可作为异相成核点,这是泡孔成核方式转变的结果。由于PE-HD分散相的引入,PLA与PE-HD之间出现了新的界面,导致了均相成核和非均相成核的共存[6]280。加入GEMA后,PLA的发泡性能进一步提高,随着GEMA含量的增加,大泡孔和小泡孔的尺寸不断减小、密度不断增加,当加到3份时小泡孔的尺寸和密度到达了16.7 μm、1.7×108个/cm3。这主要是因为,GEMA加入后PLA和PE-HD的相容性变好,熔体强度更高,PE-HD分散得更加均匀、数量更多、粒径更小。在相同的发泡条件下,泡孔成核数量更多,泡孔尺寸更小,泡孔密度更大。当GEMA含量增加到5份时,泡孔由复合泡孔转变为单一泡孔,这提供了一种新的方式来调控PLA的泡孔结构,进而调控PLA泡沫的性能。

(a)PLA (b)PLA/PE-HD (c)PLA/PE-HD/GEMA 1 (d)PLA/PE-HD/GEMA 3 (e)PLA/PE-HD/GEMA 5图6 不同PLA泡沫样品在95 ℃时泡孔形貌的SEM照片Fig.6 SEM of various PLA foams at the foaming temperature of 95 ℃
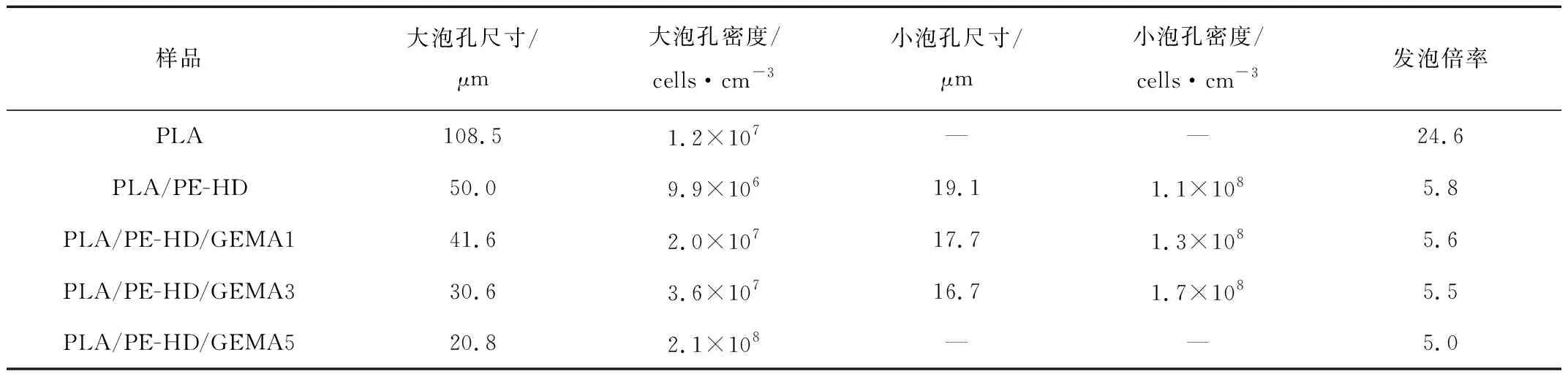
Tab.3 The cellular parameters of various PLA foams at the foaming temperature of 95 ℃
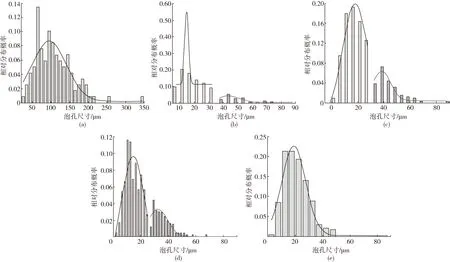
(a)PLA (b)PLA/PE-HD (c)PLA/PE-HD/GEMA 1 (d)PLA/PE-HD/GEMA 3 (e)PLA/PE-HD/GEMA 5图7 不同PLA泡沫样品在95 ℃时泡孔尺寸分布图Fig.7 Cell size distribution of various PLA foams at the foaming temperature of 95 ℃
3 结论
(1)将GEMA作为反应型增容剂,采用熔融共混的方法制备PLA/PE-HD/GEMA合金,随着GEMA含量增加,PE-HD分散相尺寸减小,PLA/PE-HD/GEMA合金的冲击强度不断提高;
(2)以CO2为发泡剂,采用间歇发泡的方法成功地制备了PLA/PE-HD/GEMA泡沫,当PLA的含量为80份,PE-HD的含量为20份,GEMA的含量为5份时所产生的泡沫的泡孔大小和泡孔密度达到20.8 μm和2.1×108个/cm3;随着GEMA含量增加,改善相容性的同时,还可以实现泡孔由复合泡孔转变为单一泡孔,这提供了一种新的方式来调控PLA的泡孔结构。
猜你喜欢
复旦学报(自然科学版)(2022年6期)2023-01-31 04:48:36
国际皮肤性病学杂志(2022年1期)2022-04-15 04:08:02
包装工程(2022年1期)2022-01-26 09:03:10
科教导刊·电子版(2021年6期)2021-05-06 05:05:14
工程塑料应用(2020年11期)2020-11-28 01:57:50
现代食品科技(2018年12期)2019-01-07 12:00:18
中国塑料(2016年6期)2016-06-27 06:34:18
合成材料老化与应用(2015年4期)2015-07-25 10:45:44
合成纤维工业(2015年4期)2015-03-25 12:52:38
科技致富向导(2013年16期)2013-09-09 01:02:58
