
NH3与BeH2反应机理的密度泛函理论研究
2019-09-17 11:36:06董肖
原子与分子物理学报 2019年4期
董 肖
(伊犁师范学院物理科学与技术学院 新疆凝聚态相变与微结构实验室,伊宁 835000)
1 引 言
氢是一种储量丰富燃烧放热量大的新型可再生清洁能源,其开发及利用能够为解决能源和环境的问题提供巨大帮助.氢能源使用过程中的关键环节就是氢的储存,安全、高效、经济的储氢技术是目前氢能规模化应用的主要瓶颈[1].2002年Chen等[2]首次提出M-N-H系(M是指I-IV族和一些过渡族金属)可作为储氢材料,主要对Li-N-H系列材料的储氢性能进行了研究.随后Chen等[3]得出Li-N-H体系材料的最大放氢量为9.6wt%,但是在放氢过程中会产生氨气.Janot等[4]发现Li-Mg-N-H体系储氢材料在放氢过程当中存在NH3的释放,NH3可与LiH迅速反应生成氢气,从而可以避免氨气所导致的污染.已有研究表明[5],储氢材料吸放氢反应过程当中所释放的氨气量与温度和氢压相关.另有研究[6]指出伴随吸放氢过程所释放的少量氨气是影响2LiNH2-MgH2/Mg(NH2)2-2LiH材料作为氢源材料在质子交换膜燃料电池中应用的关键因素之一.Luo等[7]指出2LiNH2-MgH2体系储氢材料在经过270次吸放氢循环反应之后,体系的储氢量出现25%的损失,其中1/3的损失是由于吸放氢过程氨气的形成.由此可见M-N-H体系储氢材料在储放氢反应过程中往往伴随着NH3的释放,对氢气流造成污染,对材料的储放氢性能有重要影响.另一方面,Leng等[8]指出LiH,NaH,MgH2,CaH2等氢化物可与氨气发生反应而生成氢气,且与氨气反应速度快慢的顺序为NaH,LiH,CaH2,MgH2.董肖等[9-11]对NH3与MH(M=Li,Na,Mg,Ca)的反应机理进行了理论研究.Amica等[12]发现MgH2和CaH2分别添加到LiNH2-LiH体系中能够改善体系的脱氢温度和动力学条件.可以看出碱土金属氢化物在M-N-H体系储氢材料中的重要作用.BeH2是压稳态氢化物,氢浓度高,稍微加热就会分解释放出氢气,可以作为储氢材料[13].彭敏等[14]采用第一性原理对BeH2的电子结构与光学性质进行了研究.杜泉等[15]对BeH2的分子结构和分析势能函数进行了理论计算. 而目前对BeH2与NH3反应机理的理论研究未见报道,因此本文运用量子化学密度泛函理论方法,对NH3与BeH2的反应进行了理论计算,研究其反应通道和反应机理.
2 计算方法
对团簇和化学反应机理的研究[16-18]采用量子化学方法进行计算模拟已经非常常见,并且非常适用.本文运用量子化学密度泛函理论(DFT)的B3LYP方法在6-311G(d,p)基组水平上对NH3与CaH2反应过程中各驻点的构型进行了全几何参数优化,并在相同水平上对中间体和过渡态进行了频率分析和内禀反应坐标(IRC)计算,以验证中间体与过渡态的相互连接关系和正确性.使用QCISD方法,在6-311G(d,p)基组水平上计算了反应通道上各驻点的单点能,得到相对精确的能量信息,计算各驻点的相对能量并给出反应过程的能级图.所有计算采用Gaussian 03程序完成.
3 结果与讨论
BeH2与NH3发生反应,产物为BeHNH2和H2,其中产物BeHNH2还会进一步与NH3或BeH2进行反应.计算结果表明,若第一步反应产物BeHNH2与BeH2再发生反应,也就是说BeH2和NH3反应摩尔比为2∶1时,总反应为两步氢取代反应,产物为(BeH)2NH和2H2;若第一步反应产物BeHNH2与NH3反应,即BeH2和NH3反应摩尔比为1∶2时,则总反应亦为两步氢取代反应,产物为Be(NH2)2和2H2.
3.1 NH3和BeH2的反应
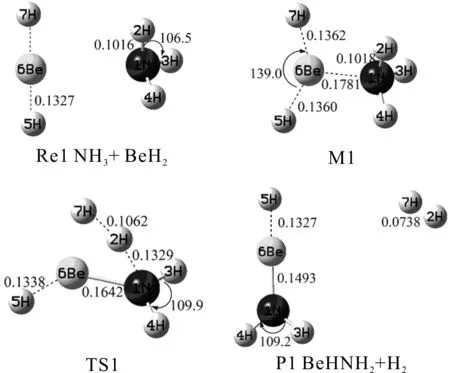
图1 NH3与BeH2反应势能面上各驻点的几何结构及参数[键长/nm,键角/(°)]Fig.1 Geometries and parameters at the critical points of the potential energy surface of the reaction between NH3 and BeH2 (bond lengths in nm,bond angles in degree)
如图1列出了BeH2与NH3反应势能面上各驻点的几何构型及具体参数.反应物Re1中,N原子进攻Be原子形成中间体M1,其中H(2)、H(3)和H(4)相对于N原子地位相同,H(5)和H(7)相对于Be原子亦存在对称性,故分析得出该氢取代反应为单通道反应.M1中的H(2)逐渐远离N(1),与H(7)原子相互靠近,形成过渡态TS1,由结果可以看出N(1)—H(2)间距有较明显增大(从0.1018增至0.1329 nm),N(1)—Be(6)间距略有减小(从0.1781到0.1642 nm).此后H(2)与H(7)进一步靠近(从0.1062到最终的0.0738 nm),N(1)—H(2)间距继续增大,形成产物P1.对TS1经过频率计算得到结果有唯一虚频,对TS1经过内禀反应坐标计算,验证了其与M1和P1驻点之间的相互连接关系的正确性.
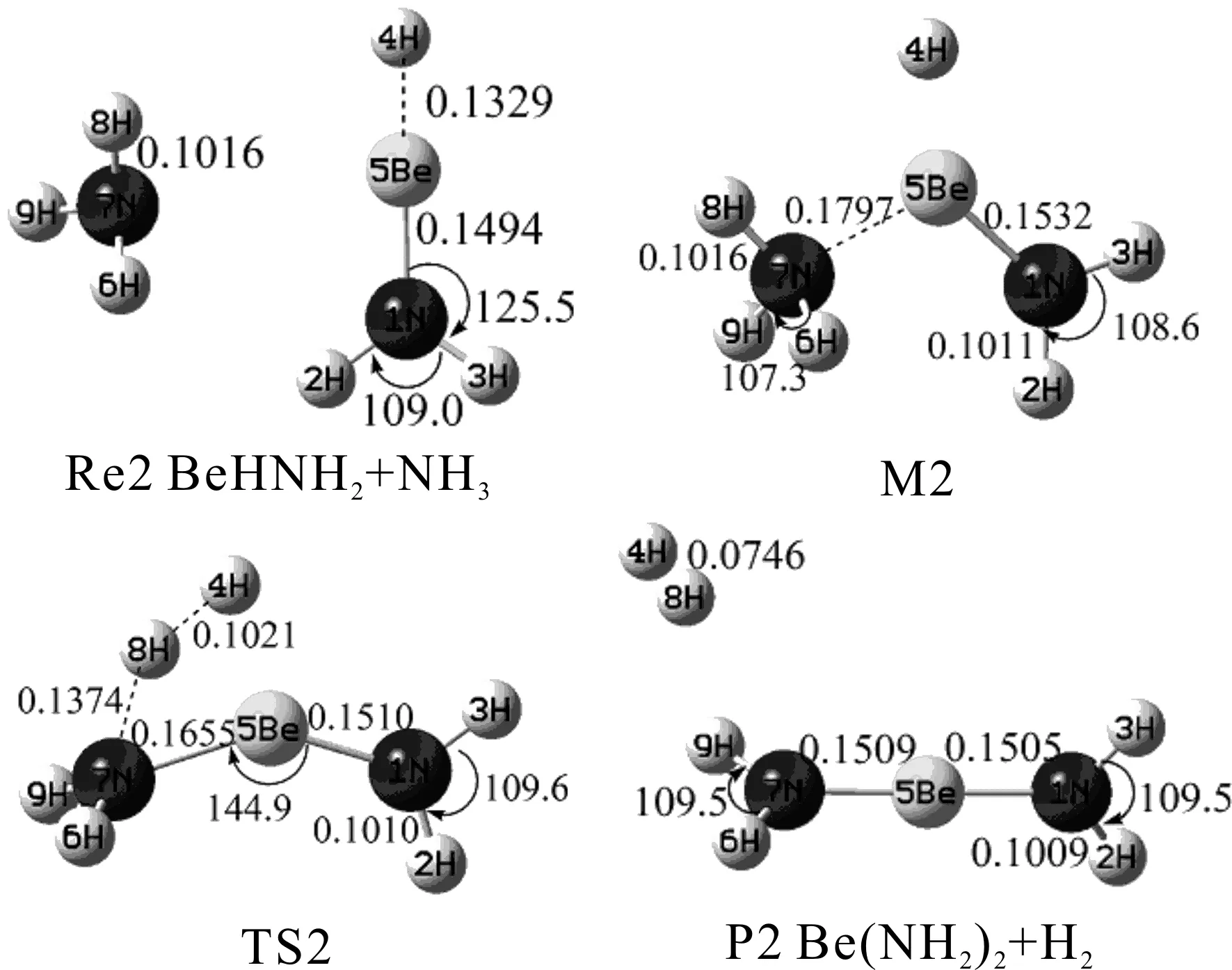
图2 BeHNH2与NH3反应势能面上各驻点的几何结构及参数[键长/nm,键角/(°)]Fig.2 Geometries and parameters at the critical points of the potential energy surface of the reaction between NH3 and BeHNH2(bond lengths in nm,bond angles in degree)
3.2 BeHNH2和NH3的反应
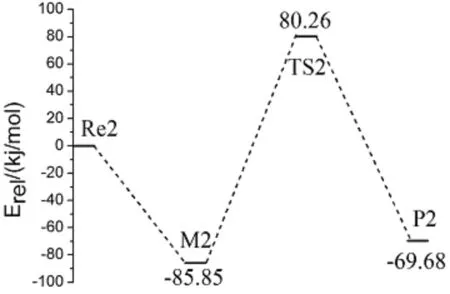
经过NH3与BeH2反应得到的产物BeHNH2与另一个NH3进一步发生反应,各驻点的几何构型及结构参数如图2所示.N(7)原子选择不同方位进攻Be(5)原子均能够形成M2同一稳定构型,在M2中N(7)与Be(5)之间距离为0.1797 nm,相比Be(5)原子与氨基官能团的距离(0.1532 nm)要大.与第一步反应过渡态的形成相类似,在M2中H(8)原子逐渐远离N(7)原子,且与H(4)原子相互靠近,形成过渡态TS2,过程中H(8)—N(7)间距有较明显增大(从0.1016增至0.1374 nm),Be(5)—N(7)间距有所减小(从0.1797减小到0.1655 nm),两N原子相对Be原子结构趋于对称.随后H(4)与H(8)进一步靠近(从0.1021到最终的0.0746 nm),N(7)—H(8)间距继续增大,形成产物P2.对TS2进行频率计算得到结果有唯一虚频,对TS2经过内禀反应坐标计算,验证了其与M2和P2驻点之间的相互连接关系的正确性.
3.3 CaHNH2和CaH2的反应
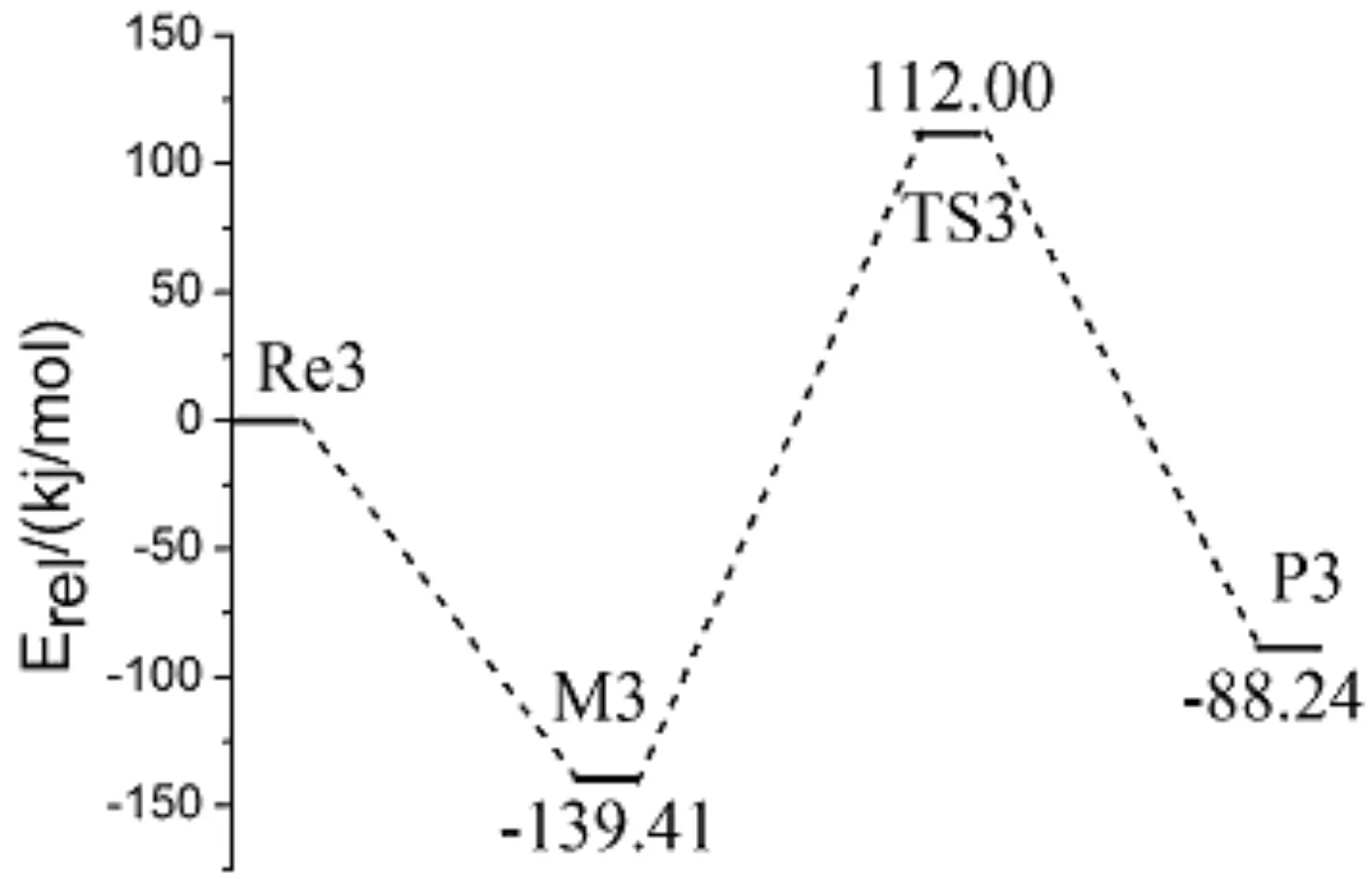
第一步反应产物BeHNH2也可与BeH2发生进一步的反应,图3给出了该反应路径上各驻点的几何构型及结构参数.反应物形成中间体过程中,Be(7)原子从不同方位靠近N原子均形成同一稳定构型的中间体M3,Be(7)与N(1)间距为0.1665 nm,Be(5)与N(1)间距为0.1664 nm,结构上相对于N(1)-H(2)-H(3)所构成平面对称. 同样,与第一步反应形成过渡态的过程相类似,在M3中H(3)原子逐渐远离N(1)原子,且与H(6)原子相互靠近,形成过渡态TS3,过程中H(3)—N(1)间距有明显增大(从0.1017增至0.1456 nm),Be(7)—N(1)间距有所减小(从0.1665到0.1540 nm),Be(5)—N(1)间距亦在减小.随后H(3)与H(6)进一步靠近(从0.1025到最终的0.0744 nm),并共同远离其他原子,形成产物P3.对过渡态的频率计算结果有唯一虚频,内禀反应坐标计算验证了其与M3和P3驻点之间的相互连接关系的正确性.

图3 BeHNH2与BeH2反应势能面上各驻点的几何结构及参数[键长/nm,键角/(°)]Fig.3 Geometries and parameters at the critical points of the potential energy surface of the reaction between BeHNH2 and BeH2 (bond lengths in nm,bond angles in degree)
3.4 反应的能量与反应机理分析
分别在B3LYP/6-311G(d,p)和QCISD/6-311G(d,p)水平下计算了各反应中驻点的总能量Etotal,给出以各反应物能量为参比的相对能量Erel以及各驻点的部分振动频率,见表1所示.图4是以相对能量Erel作出各分反应过程的势能面剖面图,以清晰反映在各分反应过程中的能垒及吸放热情况.由图四可以看出,三个分反应均为放热反应.NH3和BeH2的反应放热为78.66 kJ/mol,对应过渡态能垒为176.23 kJ/mol,此能垒与M1形成TS1过程当中N(1)—H(2)间距的增大(从0.1018增至0.1329 nm)相对应;BeHNH2与NH3反应放热为69.68 kJ/mol,对应过渡态能垒为166.11 kJ/mol,主要与M2到TS2过程当中H(8)—N(7)键的增大(从0.1016增至0.1374 nm)相对应.BeHNH2和BeH2的反应放热为88.24 kJ/mol,形成过渡态能垒为251.41 kJ/mol,主要对应H(3)—N(1)间距的增大(从0.1017增至0.1456 nm).
表1 反应各驻点的能量、相对能量及部分振动频率
Table 1 Total energies and relative energies at the critical points of potential energy surface and vibrational frequencies
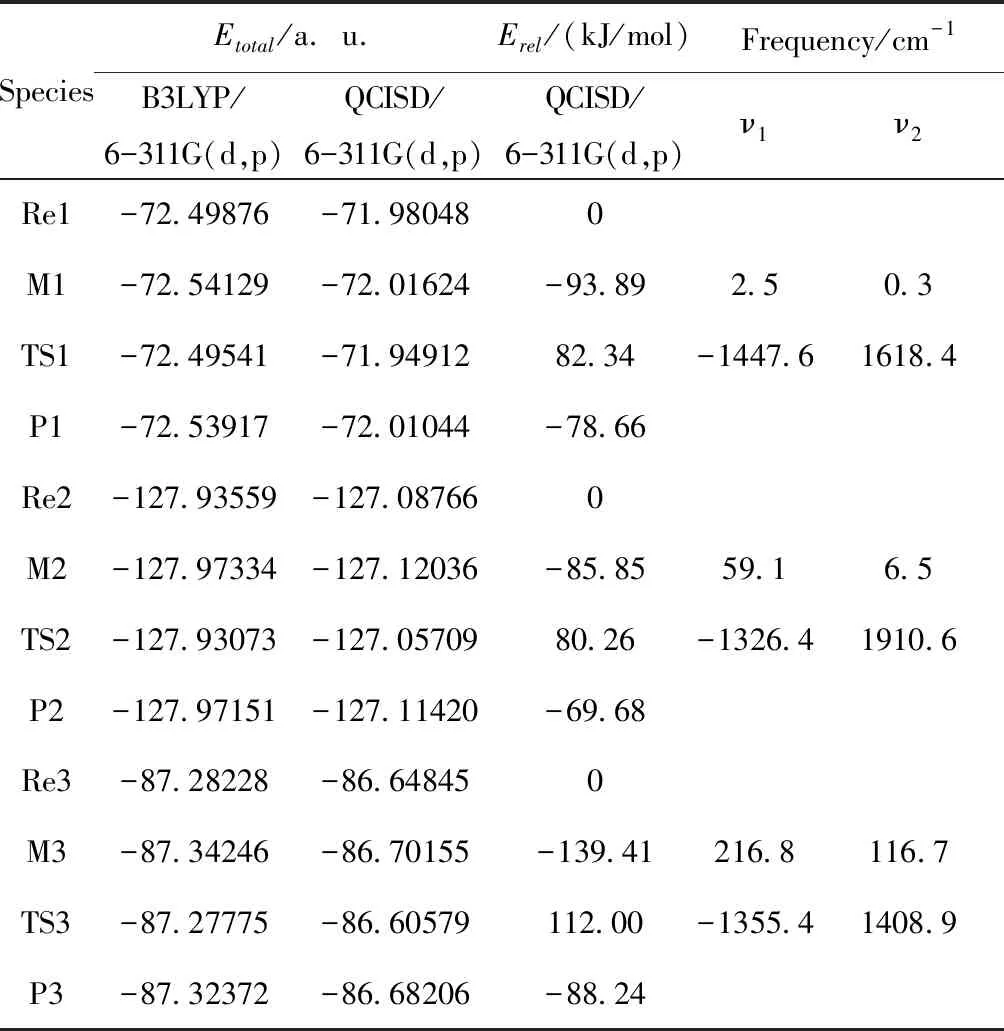
SpeciesEtotal/a.u.Erel/(kJ/mol)Frequency/cm-1B3LYP/6-311G(d,p)QCISD/6-311G(d,p)QCISD/6-311G(d,p)ν1ν2Re1-72.49876-71.980480M1-72.54129-72.01624-93.892.50.3TS1-72.49541-71.9491282.34-1447.61618.4P1-72.53917-72.01044-78.66Re2-127.93559-127.087660M2-127.97334-127.12036-85.8559.16.5TS2-127.93073-127.0570980.26-1326.41910.6P2-127.97151-127.11420-69.68Re3-87.28228-86.648450M3-87.34246-86.70155-139.41216.8116.7TS3-87.27775-86.60579112.00-1355.41408.9P3-87.32372-86.68206-88.24
对反应当中的中间体和过渡态都进行了频率计算,过渡态TS1、TS2和TS3均有且只有一个虚频,分别为-1447.6、-1326.4和-1355.4 cm-1,中间体M1、M2和M3均没有虚频,另外,通过内禀反应坐标(IRC)计算进一步确认了驻点之间的相互连接关系,最终证明了中间体与过渡态的真实性和正确性.对各反应的能垒进行比较分析,经第一步反应之后,BeHNH2和NH3的反应能垒(166.11 kJ/mol)小于BeHNH2和BeH2的反应能垒(251.41 kJ/mol),且二者相差较大,所以判断出经第一步氢取代反应之后的产物BeHNH2进一步优先与NH3发生反应.得出总体的反应过程为CaH2与NH3按照摩尔比为1∶2进行反应,产物为Be(NH2)2和2H2.反应方程式可表述为:
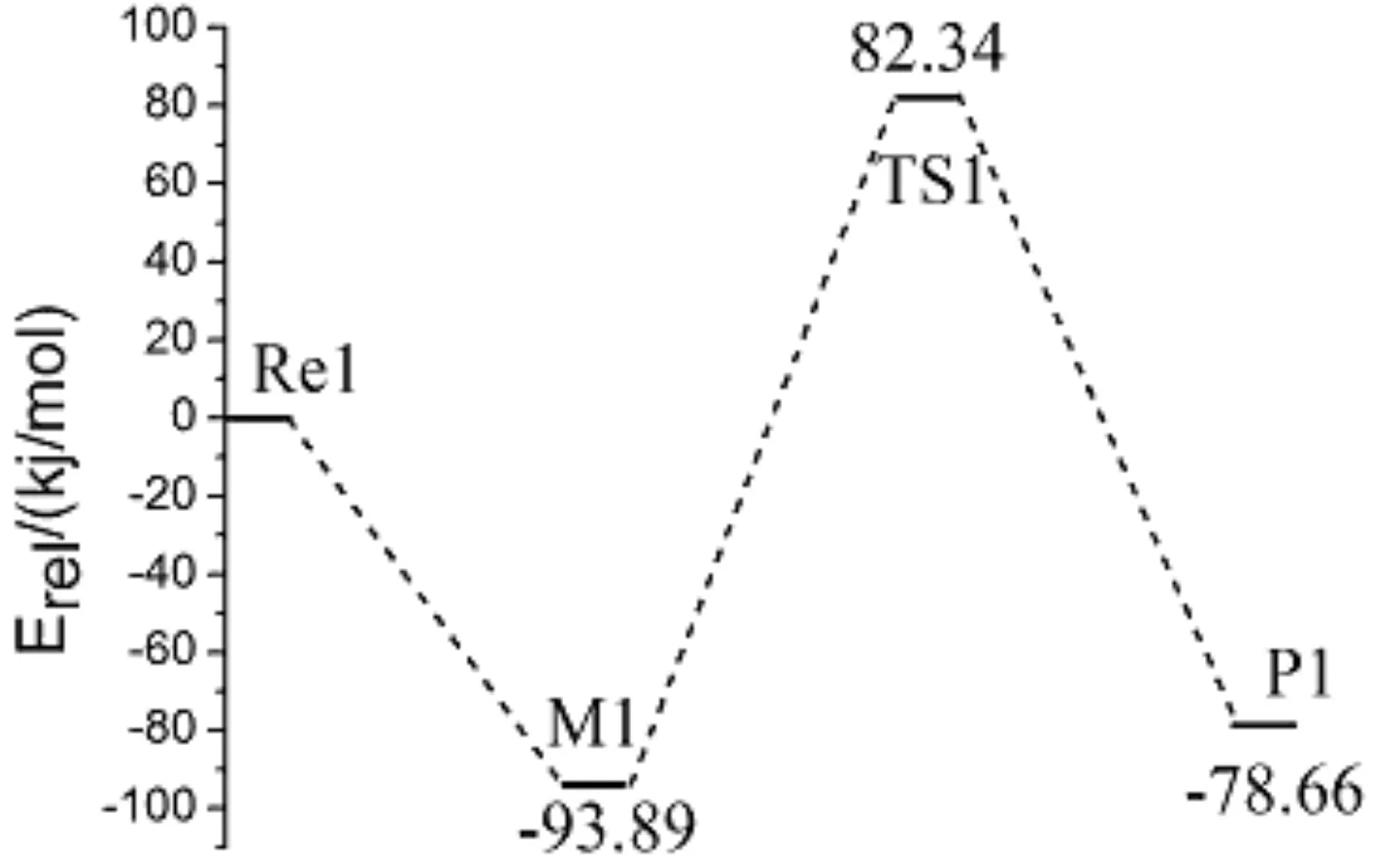
a NH3与BeH2的反应
b BeHNH2与NH3的反应
c BeHNH2与BeH2的反应
图4 各个反应过程的势能面剖面图
Fig.4 Energetic profile for potential energy surface of each reaction
BeH2+ 2NH3↔ Be(NH2)2+ 2H2
反应能垒对应于过渡态的形成,而过渡态的形成又主要与N-H键的增大相对应,结合上述反应路径分析,发现从NH3上脱氢比从-NH2上脱氢较易,同时也可以想到N-H键的断裂是反应的关键,弱化N-H键之间的作用有利于反应能垒的降低,而如何弱化N-H键的作用对于氨基类化合物储氢材料具有十分重要的意义.
另外,文献[9-11]中NH3和NaH的反应能垒为83.65 kJ/mol,NH3和LiH反应能垒为87.53 kJ/mol,NH3和CaH2的反应能垒为116.2 kJ/mol,NH3和MgH2的反应能垒为157.16 kJ/mol,得出碱土金属氢化物(LiH,NaH,MgH2,CaH2)和NH3反应速度快慢顺序为NaH,LiH,CaH2,MgH2,与文献[8]结论一致.本文经计算得出NH3和BeH2的反应能垒为176.23 kJ/mol,反应速率排在MgH2之后.
4 结 论
(1) BeH2与NH3按照摩尔比为1∶2进行反应,两步反应之后生成Be(NH2)2和2H2.
(2)过渡态的形成主要与N-H键的增大相对应,进一步弱化N—H键的作用有利于氢气的释放.相比较从NH3中脱氢比从—NH2中脱氢较容易进行.
(3)反应所释放H2中的两个H原子分别来源于NH3和BeH2.
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
中国园林(2018年7期)2018-08-07 07:07:48
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
党员电教与远程教育(2016年3期)2016-03-19 16:46:56
党的生活·党员电教与远程教育(2016年3期)2016-02-26 01:48:08
