
超高效液相色谱-串联质谱法同时分析紫薯中15种酚酸类组分
2019-09-10 07:22刘文静黄彪林香信方灵吴建鸿李巍吴妙鸿
福建农业学报 2019年10期
刘文静 黄彪 林香信 方灵 吴建鸿 李巍 吴妙鸿



摘要:【目的】运用超高效液相色谱.串联质谱(UPLC-Ms/Ms)分析不同品种紫薯中酚酸类组分及含量,为紫薯中酚酸类组分的提取、分析提供理论依据。【方法】建立超高效液相色谱.串联质谱法同时检测紫薯中15种酚酸类组分的方法,分析8种不同品种紫薯中酚酸类组分的含量。样品经甲醇冰溶液超声提取后,使用WatersT3c18色谱柱(250mm×4.6mm,5um),以0.1%(v/v)甲酸-5mmol·L乙酸铵水溶液和乙腈为流动相进行梯度洗脱,负离子扫描方式下多反应监测(MRM)模式测定。【结果】在优化的试验条件下,15种酚酸类组分在相应的浓度范围内具有良好的线性关系,相关系数(r)为0.9956~0.9997,方法检出限和定量限分别为0.3~5.8ug·L(S/N≥3)和1.0~15.2ug·L(S/N≥10】;加标试验平均回收率73.3%~97.1%,相对标准偏差1.8%~8.9%。【结论】在所分析的紫薯样品中共检出5种酚酸类组分,含量0.102~1698.490mg.kg,其中绿原酸含量最高,对香豆酸含量最低。该法灵敏、快速、准确,适用于紫薯中酚酸类组分的定性定量分析。
关键词:紫薯;酚酸类组分;超高效液相色谱-串联质谱
中图分类号:TS 215文献标志码:A 文章编号:1008-0384(2019)10-1203-08
0引言
【研究意义】紫薯(Purple sweetpotato)又名黑薯,是甘薯的一个优良新品种,其肉质部分呈紫色或者紫红色。据报道,紫薯含有多种营养功能成分:如淀粉、多糖、蛋白质、粗脂肪、多酚和花色苷等。紫薯花色苷和多酚经研究表明具有良好的抗氧化、抗肿瘤、降血压等生理功效,目前对紫薯功能性成分的研究主要集中在对紫薯花色苷成分分析,而对同样具有良好生理活性的紫薯酚酸类组分的报道则相对较少。【前人研究进展】目前报道测定植物性农产品中酚酸类组分的方法有毛细管电泳法、高效液相色谱法(highperformance liquid chromatography,HPLC)、高效液相色谱串联质谱联用法((high performance liquidchromatography-tandem mass spectrometry,HPLC-MS/MS)。毛细管电泳法灵敏度较低,高效液相色谱法灵敏度比毛细管电泳法高,但是对于基质复杂的样品分离效果不够理想,且分析时间较长。高效液相色谱串联质谱联用法是近年兴起的检测技术手段,利用离子通过不同通道技术对结构相近物质有效分离,分离时间短,灵敏度低,效果好。【本研究切入点】利用液质联用技术分析紫薯功能性成分,所需样品量少,时间短,可以同时准确检测多种组分,而利用此技术分析紫薯功能性营养功能成分的研究鲜见报道。【拟解决的关键问题】本研究以福建地区采集的8份不同品种的紫薯样品为研究对象,通过对色谱、质谱条件的优化,运用超高效液相色谱一串联质谱(UPLC-MS/MS)建立了15种酚酸类组分的检测方法,分析了不同品种紫薯中酚酸类组分及含量,旨在为紫薯中酚酸类组分的提取、分析提供理论依据,为紫薯新品种的选育与开发利用奠定基础。
1材料与方法
1.1仪器与试剂
仪器设备:UPLC H-Class超高效液相色谱仪(美国waters公司);Xevo TQ-S三重四级杆质谱(美国Waters公司);Millipore Direct-Q5超纯水仪(美国Millipore公司);SYG-2水浴恒温振荡器(常州朗越仪器有限公司);样品均质器(德国IKA公司);XW-80A旋涡混合器(上海医科大学仪器厂)。
供试紫薯样品:龙紫4号、龙紫6号、莆紫薯18号、莆紫薯3号、福宁紫3号、福宁紫4号、福薯9号、福薯24号,所有紫薯样品由福建省农业科学院作物研究所提供,采自福建省龙岩市连城县家庭农场。
试验试剂:水杨酸(salicylic acid)、对羟基苯甲酸(p-Hydroxybenzoic acid)、2,5-二羟基苯甲酸(2,5-Dihydroxybenzoic acid)、3,5二羟基苯甲酸(3,5-Dihydroxybenzoic acid)、3,4,5-三羟基苯甲酸(3,4,5-Trihydroxybenzoic acid)、绿原酸(Chlorogenic acid)、异绿原酸A(Isochlorogenic acid A)、香草酸(Vanillicacid)、咖啡酸(caffeic acid)、丁香酸(syringate)、对香豆酸(Coumalic acid)、阿魏酸(Ferulic acid)、芥子酸(sinapic acid)、2-羟基肉桂酸(2-Hydrocycinnamic-acid)、3-羟基肉桂酸等标准品(2Hydrocycirmamicacidl(纯度≥97%,南京道斯夫生物技术股份限公司);甲醇(色谱纯,美国Merck公司)、乙腈(色谱纯,美国Merck公司)、乙酸铵(LC-MS级,美国Waters公司)、甲酸(LC-MS级,美国Waters公司);试验用水为Millipore Direct-Q5超纯水仪制备,其他试剂为国产分析纯。
1.2超高效液相色谱-质谱条件
1.2.1超高效液相色谱条件Waters T3C18色谱柱(Ф1.8um,2.1mm//1×50mm),柱温:30.0%;流速:0.2mL.min;进样量:4ul;流动相A为0.1%(V/V)甲酸水溶液,流动相B为乙腈;梯度洗脱程序:0~4min,90%~10%A;4~6min,维持10%A;5~8min,10%~50%A;8~9min,50%~90%A;9~10min,维持10%A。
1.2.2质谱条件电喷雾离子化负离子(ESI-)模式;检测方式:多反应监测(MRM)模式;毛细管电压:1.5kV;锥孔气流:氮气,流速50L.h;脫溶剂温度:150°C,流速800L.h;离子源温度:150°C;雾化气压力:0.28MPa;碰撞气:高纯氩气。质谱检测参数见表1。
1.3标准溶液的配制
准确称取水杨酸、对羟基苯甲酸等15种标准品各10.0mg,分别用甲醇定容至10.00mL,得到各标准品浓度为1000mg·L的单标母液,标准品母液置于20~C冰箱中保存,试验时取母液配制成10mg·L的单标中间液,根据需要用甲醇配成合适浓度的混合标准工作储备溶液,于-20°C冰箱中保存。用于制作标准工作曲线的不同浓度的混标溶液,现配现用。
1.4样品前处理
紫薯中活性成分的提取参考蔡湛等的方法稍加改进,称取0.500g磨碎的紫薯鲜样加入5mL70%的甲醇溶液于棕色玻璃管中,70℃水浴中振荡提取30min,提取后自然冷却至室温,将提取液转移至离心管中,于5000r·rain转速下离心5min,离心结束后上清液全部转移至10mL容量瓶。残渣再加入5mL 70%甲醇溶液提取1次,重复以上操作。合并提取液定容至10.00mL,摇匀,0.45um滤膜过滤,滤液待上机分析用。
2结果与分析
2.1样品前处理条件的选择
在以往测定植物性样品的总酚报道中,多选用乙醇一水或者甲醇水为提取酚类物质的溶剂。超高效液相色谱一串联质谱的流动相体系多为甲醇一水或者乙腈一水,为检测过程中减少溶剂效应对峰型的影响,因此选择甲醇.水作为提取溶剂,试验中比较了不同浓度甲醇溶液提取效率的影响,结果表明以70%的甲醇溶液为提取剂时测得的提取液中总酚含量最高(表2),总酚测定采用稍加改进的Folin-Cioealteu方法。
2.2流动相的选择
试验中考察了以甲醇一水和乙腈一水作为流动相以及在流动相中添加甲酸、乙酸铵对目标化合物离子色谱峰峰型及响应信号的影响。试验表明:采用0.1%(V/V)甲酸.5mmol·L乙酸铵水溶液和乙腈为流动相,在负离子扫描模式下,酚酸类组分有良好的峰形且相对于不加乙酸铵的流动相有更好的响应强度。所分析的酚酸类组分,大都具有酚羟基官能团或同时含有羟基与羧基结构,表现出一定的弱酸性。在流动相中加入乙酸铵,有利于所分析的目标化合物中氢离子的解离,形成目标负离子,增强响应信号;而甲酸的加入有利于抑制酚酸类组分与色谱柱的静电作用,减少拖尾现象,改善峰形。因此,试验中采用0.1%(v/v)甲酸-5mmol.L乙酸铵水溶液和乙腈作为流动相。
2.3质谱条件的优化
Waters三重四极杆质谱条件的优化主要包括选择各化合物的母离子、子离子、锥孔电压(CV)、碰撞能量(cE)等参数的确定。配制500.0ug·L的15种酚酸类组分单标溶液,流动相为0.1%(V/V)甲酸.5mm01.L乙酸铵水溶液:流动相B(乙腈)=50%:50%,经质谱直接进标准溶液,采用电喷雾正离子模式(ESI)和负离子模式(ESr)通过自动调谐的方式分别对各化合物单标溶液进行母离子、子离子扫描。试验发现相对于正离子扫描模式,在负离子扫描模式下更容易得到稳定的母离子及子离子,响应信号更好。
2.4色谱柱的选择
试验中比较了分别使用Waters BEH C18柱和Waters T3C18柱,在选用甲醇.水和乙腈.水不同流动相体系对15种酚酸类组分的保留和分离情况。发现Waters T3C18对极性的酚酸类组分的分离效果更好,具有更窄的峰型,尤其是对于对羟基苯甲酸及3,5-二羟基苯甲酸等化合物,区别更为明显。窄的峰型更有利于分析中的准确定量,因此在试验中选用Waters T3C18作为分离柱。
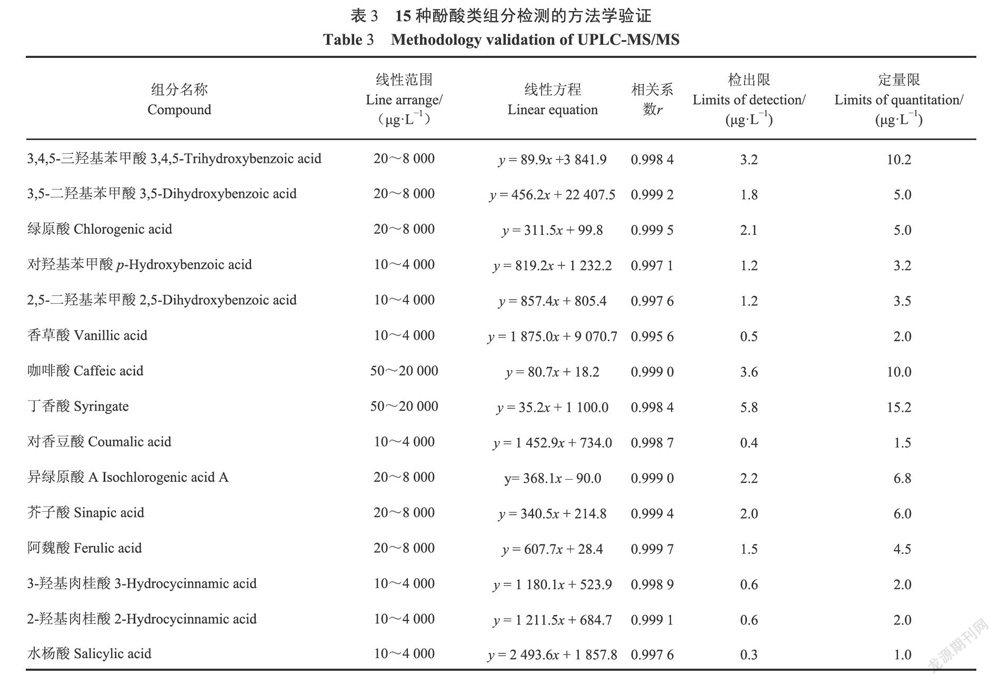
2.5方法评价
在优化的条件下,考察了15种目标化合物的线性范围、相关系数及在基质中的加标回收率情况。结果显示,在相应的浓度范围内,目标化合物的标准曲线标准溶液浓度(x,ug·L)与对应的峰面积∽呈良好线性关系,相关系数(r)为0.9956~0.9997。分别依据特征离子色谱峰色谱信号的3倍信噪比(S/N≥3)和10倍的信噪比确定方法的检出限和定量限(S/N≥10),方法的检出下限为0.5ug·L~5.8ug·L和定量限分别为1.0ug·L~15.2ug·L(表3),能满足定量分析的要求。
根据样品中各酚酸类组分的含量情况,根据各化合物的线性范围,确定在样品中合适的加标量。对未检出的组分在定量限水平参考GB/T 27404中要求,按照优化的试验条件测定,进行低、中、高水平的加标,重复6次。样品加标回收试验平均回收率76.3%~96.8%,相对标准偏差0.6%~7.9%,分析方法准确度较好(表4)。
2.6不同紫薯样品中活性成分含量的检测分析
收集福建产区8种不同品种的紫薯样品为试验材料,按1.3所述样品前处理方法得样品待测溶液,运用所建立的方法对样品待测液中的酚酸类组分含量进行检测,进样量为4ul。每个样品重复测定3次。在所收集的紫薯样品中共检出5种酚酸类组分,分别为香草酸、咖啡酸、阿魏酸、绿原酸、异绿原酸A。紫薯样品中酚酸类组分含量为0.102~1698.490mg·kg,具体含量见表5。结果表明:在紫薯样品中含量最高的为绿原酸,含量范围291.439~1698.490mg·kg,绿原酸的含量范围与徐柯等报道的较为接近;其次为异绿原酸A;含量最低的为对香豆酸。
3讨论与结论
通过对色谱.质谱条件的优化,建立了超高效液相色谱一串联质谱法同时分析紫薯中15种酚酸类组分检测方法。收集了福建产区8份不同品种的紫薯样品,运用所建立的方法进行检测。在所分析的紫薯样品中共检出5种酚酸类组分,含量在0.102~1698.490mg'kg,在所有的紫薯样品中,相对于其他酚酸组分,绿原酸的含量均为最高,最高含量为1698.490mg·kg,最低含量为291.439mg·kg;其次为异绿原酸A,最高含量为202.682mg.kg,最低含量为34.099mg;对香豆酸含量为最低。因为品种的差别,各酚酸类组分的含量表現出一定的差异,在所有的样品中,福宁紫3号紫薯样品中,酚酸的含量为最高。建立的方法灵敏、快速、准确,适用于紫薯中酚酸类组分的定性定量分析,可为紫薯的资源开发利用提供参考依据。
猜你喜欢
阅读(科学探秘)(2021年3期)2021-06-02
家教世界·V家长(2020年10期)2020-11-20
中国中药杂志(2016年21期)2017-02-16
中国中药杂志(2016年21期)2017-02-16
分析化学(2017年1期)2017-02-06
分析化学(2017年1期)2017-02-06
热带农业科学(2016年10期)2016-12-12
红领巾·萌芽(2016年1期)2016-09-10
上海医药(2016年1期)2016-02-22
安徽农学通报(2015年10期)2015-06-15
