
超高效液相色谱—串联质谱法快速测定猪尿液中30种不同种类“瘦肉精”药物残留
2017-02-06 21:34
分析化学 2017年1期


摘 要 建立了超高效液相色谱.串联质谱快速简单地同时测定猪尿液中30种不同种类“瘦肉精”药物(赛庚啶、可乐定及28种β.受体激动剂类)残留的方法。对液相色谱分离条件、MS/MS检测参数及样品前处理方式进行了优化。试样经5000 r/min离心5 min后直接经MCX柱净化,分别用3 mL水和3 mL甲醇淋洗,5%氨化甲醇进行洗脱,N2吹干后以流动相进行复溶,UPLC.MS/MS进行测定。结果表明,30种药物可在5.0 min内有效分离; 各药物在0.1~10 μg/L范围内呈良好的线性关系,相关系数均大于0.992; 方法的检出限为0.1 μg/L,定量限为0.3 μg/L。在3个浓度水平下的平均回收率为67.6%~103.2%,日内、日间相对标准偏差分别为2.8%~16.8%和2.6%~15.8%。
关键词 超高效液相色谱.串联质谱法; 痕量残留; 猪尿液; 瘦肉精
1 引 言
“瘦肉精”(Lean meat agents)最早是盐酸克伦特罗的俗称,现在泛指一类可以促进动物生长,提高瘦肉率的药物[1],主要包括盐酸克伦特罗、沙丁胺醇、莱克多巴胺、苯乙醇胺A、氯丙那林等β.受体激动剂,以及近年来监测发现的赛庚啶、可乐定等具有促进动物生长、提高饲料转化率的化合物[2]。由于上述药物在动物性食品中的极易残留,会对食用者产生系列毒副作用,因此,包括我国在内的许多国家和国际组织已经制定了极其严格的规定。到目前为止,我国已明确禁止在食品动物养殖过程中使用克伦特罗、沙丁胺醇、莱克多巴胺、苯乙醇胺A、氯丙那林、多巴胺、班布特罗、齐帕特罗、马布特罗、西马特罗、溴布特罗、福莫特罗/阿福特罗、特布他林、赛庚啶、可乐定共15种具有促进动物生长、提高瘦肉率的药物[1]。
近年来,我国加大了对明令禁止的上述15种“瘦肉精”非法使用的打击力度,“瘦肉精”药物在畜禽养殖中的非法使用得到了有效遏制,然而具有促进动物生长、提高瘦肉率功效、而未被列入国家禁用药物名单的β.受体激动剂同系物或衍生物(如克罗潘特、羟甲基克伦特罗、克伦丙罗、西布特罗、妥布特罗、马喷特罗、喷布特罗、卡布特罗、丁酚胺、利托君、克伦己罗、克伦塞罗、异克伦潘特、溴氯特罗、苯氧丙酚胺等)极易被不法分子利用,作为新型“瘦肉精”替代物,用于生猪养殖过程中,达到非法牟利的目的,进而威胁广大消费者的健康安全。
尿液常被选择作为药物代谢、残留消除实验的重要靶组织,而且,与其它组织样品相比,尿液在样品采集过程中可有效避免对动物造成损伤,因此,动物尿液通常作为大批量样品监测的测试对象[3]。近年来,研究者针对动物尿液中β.受体激动剂药物残留建立了包括液相色谱[4]、气相色谱.串联质谱[5]、液相色谱.串联质谱[3,6~11]、ELISA[12]、胶体金试纸法[13]、免疫传感器[14]、液相芯片法[15]等一系列快速检测及定量确证检测方法。而赛庚啶、可乐定作为新型“瘦肉精”药物,分别属于H1受体拮抗药和α2.受体激动剂[14,15],其化学结构与β.受体激动剂药物有较大的差异(各药物的化学结构见图1),理化特性、色谱行为也有较大差别,因而同时检测赛庚啶、可乐定与β.受体激动剂的方法鲜有报道[16]。
本研究建立了超高效液相色谱.串联质谱快速、简单地同时测定猪尿液中30种不同种类“瘦肉精”药物(赛庚啶、可乐定及28种β.受体激动剂类)残留的方法,为加强生猪养殖、屠宰过程中违法使用不同种类“瘦肉精”的监管提供了可靠的技术支撑。
2 实验部分
2.1 仪器与试剂
AcquityTM UPLC.Xevo TQ MS超高效液相色谱.串联质谱仪(配电喷雾离子源)、UPLC CSH C 18色谱柱(100 mm×2.1 mm,1.7 μm)、BEH C 18(100 mm×2.1 mm,1.7 μm)、HSS T3(100 mm×2.1 mm,1.7 μm)、BEH Shield RP 18(100 mm×2.1 mm,1.7 μm)、固相萃取装置、Oasis MCX固相萃取柱(60 mg, 3 mL)均为美国Waters公司产品; 5804R高速冷冻离心机(德国Eppendorf公司); 氮吹仪(美国Organomation公司)。
标准品: 盐酸克仑特罗(Clenbuterol hydrochloride, 98.5%)、盐酸溴布特罗(Brombuterol hydrochloride, 98.5%)、盐酸班布特罗(Bambuterol hydrochloride, 98.0%)、莱克多巴胺(Ractopamine, 97.0%)、沙丁胺醇(Salbutamol, 100%)、盐酸克伦潘特(Clenpenterol hydrochloride, 99.5%)、羟甲基克伦特罗(Hydroxy.methyl clenbuterol, 99.0%)、克伦丙罗(Clenproperol, 99.0%)、特布他林(Terbutaline, 985%)、非诺特罗(Fenoterol, 100 mg/L, 溶于乙腈)购自Dr. Ehrenstorfer GmbH公司; 苯乙醇胺A(Phenylethanolamine A, 98.0%)、盐酸马布特罗(Mabuterol hydrochloride, 99.0%)、齐帕特罗(Zilpaterol, 98.0%)、卡布特罗半硫酸盐(Carbuterol Hemisulfate Salt, 98.0%)、硫酸丁酚胺(Bamethane sulfate, 98.0%)、苯氧丙酚胺(Isoxsuprine, 98.0%)购自Toronto Research chemicals INC公司; 盐酸利托君(Ritodrine, 98.0%)购自Tokyo Chemical Industry公司; 盐酸可乐定(Clonidine hydrochloride)、盐酸赛庚啶(Cyproheptadine hydrochloride)购自European Pharmacopoeia公司; 氯丙那林(Clorprenaline, 99.0%)、西布特罗(Cimbuterol, 99.0%)、西马特罗(Cimaterol, 99.0%)、妥布特罗(Tulobuterol, 99.0%)、克伦己罗(Clenhexerol, 99.6%)、盐酸克伦塞罗(Clencyclohexerol hydrochloride, 99.0%)、盐酸异克伦潘特(Clenisopenterol hydrochloride, 99.7%)、盐酸马喷特罗(Mapenterol hydrochloride, 99.0%)、盐酸溴氯特罗(Bromchlorbuterol hydrochloride, 99.0%)、盐酸喷布特罗(Penbutolol hydrochloride, 99.0%)购自Witega Laboratorien BerlinAdlershof GmbH公司; 酒石酸福莫特罗 (Formoterol tartrate, 98.0%)购自美国International Laboratory 公司。
2.2 标准溶液配制
分别准确称取各标准品(10.00±0.10)mg, 以甲醇溶解并定容至10 mL, 即为各标准物质储备液。分别量取适量的30种药物储备液于另一洁净10 mL容量瓶中混合,配制100 μg/L混合标准工作液, 20℃保存备用。
2.3 样品前处理
准确量取尿液5.00 mL于50 mL具塞离心管中,5000 r/min离心5 min,将上清液用5%乙酸溶液调至pH 3.0后,加入经3 mL甲醇、3 mL水活化的MCX柱,分别用3 mL水和3 mL甲醇洗涤,真空泵抽干后以3 mL 5%氨化甲醇洗脱,45℃水浴条件下N2吹近干,用1 mL 0.1%甲酸.乙腈(9∶1, V/V)溶液复溶,UPLC.MS/MS检测。
2.4 色谱.质谱条件
色谱条件: 流动相A为0.1%甲酸溶液,B为乙腈,液相洗脱梯度程序: 0~1.0 min,95% A; 1.0~3.0 min,95%~87% A; 3.0~4.0 min,87%~60% A; 4.0~5.0 min,60%~10% A; 5.0~5.1 min,10%~95% A; 5.1~7.0 min,95% A。进样量: 5 μL,柱温: 35℃,流速: 0.3 mL/min。
质谱条件: 电喷雾离子源(ESI),正离子扫描,多反应监测(MRM)模式。毛细管电压2700 V,离子源温度150℃,雾化室温度450℃,去溶剂气(氮气)流量: 1000 L/h; 锥孔气(氮气)流量: 50 L/h; 碰撞气(氩气)流量: 0.24 L/h。
3 结果与讨论
3.1 质谱参数的确定
选择一定浓度的标准溶液,利用Intelstart软件,以ESI正离子模式对待测药物进行母离子确认及子离子全扫描。确定以[M+H]+作为各药物的分子离子,分别对仪器的锥孔电压、碰撞能量等参数进行优化,进行子离子扫描,选取2个丰度强、稳定的碎片离子作为定性与定量离子,以满足欧盟2002/657/EC[17]对于禁用药物LC.MS/MS方法的定性监测的规定,即满足1个母离子、2个子离子共4个鉴定点数的要求。因此,本方法根据待测药物的保留时间,采用分段扫描的方式对不同药物的特征离子进行数据采集,以提高灵敏度。30种药物的保留时间及定性、定量离子等参数见表1。
3.2 色谱条件的优化
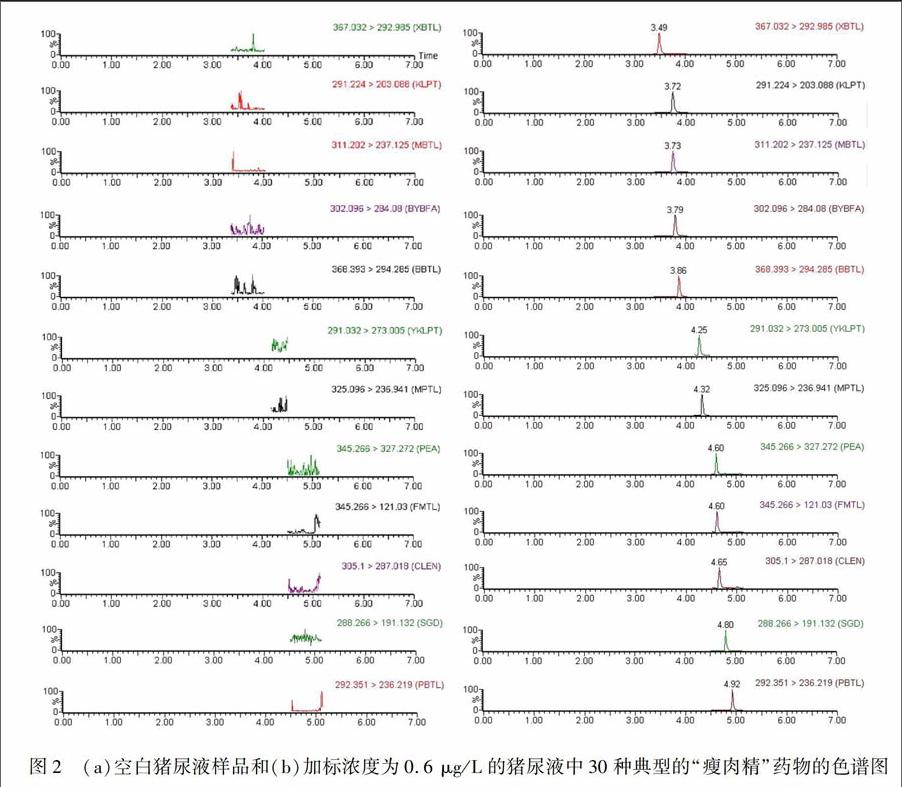
由于28种β.受体激动剂药物具有较为相似的化学结构及相近的色谱保留特性,而可乐定和赛庚啶与β.受体激动剂药物化学性质、色谱行为差异较大[16],为了将上述药物在尽可能短的时间内有效分离,本实验分别考察了BEH C 18(100 mm×2.1 mm,1.7 μm)、HSS T3(100 mm×2.1 mm,1.7 μm)、CSH C 18(100 mm×2.1 mm,1.7 μm)、BEH Shield RP 18(100 mm×2.1 mm,1.7 μm)4种型号的色谱柱对30种目标化合物的分离效果。结果表明,HSS T3色谱柱对目标化合物的保留能力较弱,尤其是沙丁胺醇、西马特罗、特布他林等12种极性较强的药物,即使用含量低于5% 的乙腈进行淋洗,其保留时间均小于1.0 min, 且不能有效分离。其余3种色谱柱均能有效保留30种药物,而采用CSH C 18色谱柱较BEH C 18和Shield RP 18色谱柱,可有效分离上述药物,且各化合物峰形尖锐。图2为30种典型的瘦肉精的空白和加标样品色谱图。
为了更好地将待测物有效分离,降低共流出物基质干扰,本实验还考察了甲醇、乙腈分别与水、0.1%甲酸及含 50 mmol/L甲酸铵的0.1%甲酸溶液等作为流动相,对目标化合物的峰形、分离度、灵敏度等的影响。结果表明,流动相中添加甲酸后,待测物的信号强度明显提高,且药物保留时间有所提前; 而以50 mmol/L甲酸铵.0.1%甲酸溶液作为流动相时,待测物的信号强度、分离度等与0.1%甲酸溶液无显著差异。甲醇和乙腈作为流动相对信号强度的影响不显著,而甲醇作为流动相进行梯度洗脱时,色谱柱压差变化范围较大,平衡时间延长。因此,本实验采用UPLC CSH C 18(100 mm×2.1 mm,1.7 μm)作为分析柱,以乙腈.0.1%甲酸作为流动相进行梯度洗脱。30种“瘦肉精”药物可在5.0 min内实现良好分离,各药物的定量离子的提取离子色谱图如图2所示。
3.3 样品前处理条件的优化
本方法采用将尿液简单离心,直接用MCX固相萃取柱净化的样品前处理方法。为了充分确保本方法的可靠性,本实验考察了文献[5,19~21]所采用的样品前处理方法(需酶解、液液萃取和/或固相萃取等步骤),分别采用叔丁醇.叔丁基甲醚[19]、乙酸乙酯/异丙醇[5,20]、HClO4[20,21]等试剂对猪尿样品中待测药物的提取,对各操作方法的提取操作步骤、所需的主要化学试剂及数量、方法的回收率、精确度等技术参数进行比较。检测结果表明,本方法不仅可获得较为满意的方法准确度、精确度(如表2所示,本实验采用尿液经离心后直接净化,30种药物的回收率在67.6%~103.2%,相对标准偏差为2.8%~16.8%),满足药物残留定量确证检测的要求,而且操作步骤简单、快速,试剂使用量少,不需要复杂的样品前处理步骤,平均每个样品的提取、净化过程仅使用很少量的有机溶剂(约7 mL甲醇),较文献[5,19~21]大大减少了有机溶剂的使用量,尤其是高氯酸等卤素试剂的使用量。本方法可作为环境友好型的样品前处理方法,适用于猪尿液样品中不同种类“瘦肉精”多残留大批量样品的筛选及确证检测。
3.4 方法学考察
3.4.1 基质添加标准曲线、线性范围、相关系数、检出限和定量限 在上述最佳色谱.质谱条件下,将空白猪尿样品按照2.3节进行样品处理,在洗脱液中加入一系列30种药物混合标准溶液(0.1, 0.3, 0.5, 1.0, 2.0, 5.0和10.0 μg/L),LC.MS/MS进行检测,以各组分的浓度与其定量离子峰面积进行线性回归,呈良好的线性关系(r2>0.992)。以3倍信噪比(S/N)计算,本方法测定猪尿液中30种“瘦肉精”药物的检出限为0.1 μg/L; 以l0倍信噪比(S/N)计算,定量限为0.3 μg/L。
3.4.2 准确度和精密度 本实验,分别在猪尿液中以3个浓度水平(0.3, 0.6和3.0 μg/L)进行空白样品加标回收实验,考察方法的回收率、日内变异系数和日间变异系数,以基质添加标准曲线进行校正,结果如表2所示。30种“瘦肉精”药物的平均回收率在67.6%~103.2%之间,日内、日间相对标准偏差分别为2.8%~16.8%和2.6%~15.8%。
3.5 实际样品的检测
运用本方法对浙江省某生猪屠宰场随机抽取的100份猪尿液进行测定,与农业部1025号公告.11.2008[5]和农业部1063号公告.3.2008[19]两个标准方法的测定结果基本一致,有1份样品检出莱克多巴胺残留,其余样品均未检出“瘦肉精”药物残留。利用本方法及两个行业标准测定的检出值分别为6.58、7.03和6.46 μg/L,变异系数小于4.5%; 而本方法却极大地节省了检测时间, 有机溶剂使用较少, 说明本方法简单快速、灵敏、可靠。
References
1 ZHAO Si.Jun, ZHENG Zeng.Ren, QU Zhi.Na, XIAO Xiao, WANG Yu.Dong, WANG Juan, LU Ping. China Animal Health Inspection, 2011, 28(4): 1-4
赵思俊, 郑增忍, 曲志娜, 肖 肖, 王玉东, 王 娟, 路 平. 中国动物检疫, 2011, 28(4): 1-4
2 LI Dan.Ni, ZHANG Wen.Gang, YAN Feng, GU Xin. China J. Veterin. Drug, 2011, 45(10): 20-24
李丹妮, 张文刚, 严 凤, 顾 欣. 中国兽药杂志, 2011, 45(10): 20-24
3 LI Xiao.Min, GAO Yan, SU Xiao.Ou, LI Yang, WANG Rui.Guo, ZHANG Yu, FU Jian.Jie. Chinese J. Anal. Chem., 2014, 42(12): 1179-1184
李晓敏, 高 燕, 苏晓鸥, 李 阳, 王瑞国, 张 瑜, 傅建捷. 分析化学, 2014, 42(12): 1179-1184
4 Du W, Zhao G, Fu Q, Sun M, Zhou H Y, Chang C. Food Chem., 2014, 145: 789-795
5 Determination of β.Agonists Residues in Pig Urine by Liquid Chromatography.Tandem Mass Spectrometry. No. 1025.11.2008 Bullentin of the Ministry of Agricluture, National Standards of the People′s Repulic of China
猪尿中β.受体激动剂多残留检测 液相色谱串联质谱法. 农业部1025号公告.11.2008, 中华人民共和国国家标准
6 WANG Pei.Long, FAN Li, SU Xiao.Ou, YE Zhi.Hua. Chinese J. Anal. Chem., 2012, 40(3): 470-473
王培龙, 范 理, 苏晓鸥, 叶志华. 分析化学, 2012, 40(3): 470-473
7 GU Xu, LIU Yi.Ming, YAO Ting, SHI Hua.Le, LI Jun, ZHAO Zhen, QIN Yu.Chang. Chinese J. Anal. Chem., 2014, 42(11): 1692-1696
谷 旭, 刘义明, 姚 婷, 石华乐, 李 俊, 赵 祯, 秦玉昌. 分析化学, 2014, 42(11): 1692-1696
8 Mauro D, Ciardullo S, Civitareale C, Fiori M, Pastorelli A A, Stacchini P, Palleschi G. Microchem. J., 2014, 115: 70-77
9 Li T T, Cao J J, Li Z, Wang X, He P L. Food Chem., 2016, 192: 188-196
10 González.Antna, Domínguez.Romero J C, García.Reyes J F, Rodríguez.González H, Centineo G, Alonso J I G, Molina.Díaz A. J. Chromatogr. A, 2013, 1288: 40-47
11 Sheu S Y, Lei Y C, Tai Y T, Chang T H, Kuo T F. Anal. Chim. Acta, 2009, 654: 148-153
12 Song C M, Zhi A M, Liu Q T, Yang J F, Jia G C, Shervin J, Tang L, Hu X F, Deng R G, Xu C L, Zhang G P. Biosensors and Bioelectronics, 2013, 50: 62-65
13 LIANG Shi.Zheng, PAN Jia.Rong, ZHANG Chi, FENG Tao, FU Li.Li. Chinese J. Anal. Chem., 2016, 44(4): 640-646
梁世正, 潘家荣, 张 弛, 冯 涛, 傅丽丽. 分析化学, 2016, 44(4): 640-646
14 ZHAN Pan, DU Xiao.Wen, GAN Ning, LIN Sai.Chai, LI Tian.Hua, CAO Yu.Ting, SANG Wei.Guo. Chinese J. Anal. Chem., 2013, 41(6): 828-834
詹 盼, 杜晓雯, 干 宁, 林赛钗, 李天华, 曹玉廷, 桑卫国. 分析化学, 2013, 41(6): 828-834
15 Müller C, Ramic M, Harlfinger S, Hünseler C, Theisohn M, Roth B. J. Chromatogr. A, 2007, 1139: 221-227
16 El.Gindy A, El.Yazby F, Mostafa A, Maher M M. J. Pharm. Biomed. Anal., 2009, 50: 1044-1049
17 Zhang G J, Fang B H, Liu Y H, Wang X F, Xu L X, Zhang Y P, He L M. J. Chromatogr. B, 2013, 936: 10-17
18 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (2002/657/EC). Off. J. Eur. Communities, 2002, L221: 8-36
19 Determination of β.Receptor Agonists in Animal Urine Liquid Chromatography.Tandem Mass Spectrometry. No. 1063.3.2008 Bullentin of the Ministry of Agricluture, National Standards of the People′s Repulic of China
动物尿液中11种β.受体激动剂的检测 液相色谱串联质谱法. 农业部1063号公告.3.2008, 中华人民共和国国家标准
20 CHEN Qi.Huang. Fujian J. Agricultural Sci., 2012, 27(6): 596-600
陈其煌. 福建农业学报, 2012, 27(6): 596-600
21 FAN Sai, ZOU Jian.Hong, MIAO Hong, WU Yong.Ning, ZHAO Yun.Feng. Chinese J. Anal. Chem., 2011, 39(8): 1153-1158
范 赛, 邹建宏, 苗 虹, 吴永宁, 赵云峰. 分析化学, 2011, 39(8): 1153-1158
Abstract An ultra.high performance liquid chromatography.tandem mass spectrometric (UPLC.MS/MS) method for simultaneous determination of thirty different species of 'lean meat agents' (including clonidine, cyproheptadine, and other 28 β.agonists) residues in swine urine was developed. The parameters of separation conditions of liquid chromatography, MS/MS detection parameters and sample preparation procedure were optimized. After centrifuged at 5000 r/min for 5 min, the swine urine samples were performed directly by MCX column. Then the column was washed with 3 mL of water and 3 mL of methanol, respectively, and then the compounds were eluted with 5% ammonium hydroxide in methanol. The analytes were detected by UPLC.MS/MS after the elution was evaporated to dryness at 45℃ under a stream of nitrogen gas. Under the optimum conditions, thirty of analytes could be well separated in less than 5 min. The linear ranges were 0.1-10 μg/L for all analytes with the correlation coefficient (r2) higher than 0.992. Limits of detection (LOD) and limits of quantification (LOQ) of this method were less than 0.1 μg/L and 0.3 μg/L, respectively. The mean recoveries of three spiked concentration levels in blank sample varied from 67.6% to 103.2% with the intra. and inter.relative standard deviations of 2.8% to 16.8% and 2.6% to 15.8%, respectively. Overall, the proposed method is simple, quick, reliable, sensitivity, and can be applied in large scale supervision of illegal usage of 30 kinds of 'lean meat agents′.
Keywords Ultra.high performance liquid chromatography.tandem mass spectrometry; Trace residues; Swine urine; Lean meat agents
猜你喜欢
杂文选刊(2021年5期)2021-05-17
食品安全导刊(2018年6期)2018-08-20
饮食与健康·下旬刊(2018年1期)2018-04-11
河北渔业(2017年4期)2017-05-26
中国纤检(2016年12期)2017-01-20
上海医药(2016年1期)2016-02-22
安徽农学通报(2015年10期)2015-06-15
分析化学(2014年3期)2014-12-16
分析化学(2014年3期)2014-12-16
现代仪器与医疗(2014年1期)2014-03-28
