
自噬—脓毒症的潜在治疗途径
2019-07-16 07:56李雯练睿沈美佳薛云允孙力超
中国继续医学教育 2019年19期
李雯 练睿 沈美佳 薛云允 孙力超
脓毒症(sepsis)是一种失调的宿主免疫反应,是由病原体感染引起的复杂炎症反应综合征[1]。自噬可以认为是机体细胞自我保护的一种方式,其程序化地对细胞器及蛋白质进行降解,在酵母中,自噬被认为是对饥饿的生理反应,以维持营养供应,在此期间,细胞质内容物被溶酶体非选择性的分离,并作为基本的代谢基质循环;同时,错误折叠的蛋白质和过剩细胞器将会被不同类型的选择性自噬进行降解。自噬活性的上调与炎症反应、免疫细胞的存活和功能有关,其活性升高能对改善器官功能障碍起到帮助作用,表现为自噬空泡的增加和自噬相关蛋白表达的增加。然而,尽管自噬通常被认为是有益的细胞保护机制,过高或过低程度的自噬都对机体不利,如果自噬超过正常水平,它会导致细胞死亡。现对自噬在脓毒症方面的研究进行综述。
1 自噬的过程
自噬通过诱导、形成自噬体、转运融合和降解发生[2],进行分解代谢,如图1所示。也就是说,在诱导自噬的信号发出并被接收后,细胞质蛋白、细胞器或病原体的囊泡就会被分离出来,形成自噬体的双膜囊泡,接着自噬体和溶酶体进行融合,形成自噬溶酶体,随后所包含的内容物将连同内膜一起被降解,自噬体外膜也脱落并循环,残渣留在胞浆中或被排出体外[3-4]。通过双膜噬菌体成熟成自噬体然后与溶酶体融合,非选择性的隔离胞质物质的途径称为非特异性途径,此外,需要受体蛋白来识别特定的物质进行降解的途径称为靶向自噬途径,靶向自噬途径在消除微生物、回收受损的线粒体方面尤为重要。
在正常的生理机制下,自噬根据机体的需求发生以维持正常细胞稳态。目前认为,自噬的调控因子有雷帕霉素靶蛋白TOR激酶、真核起始因子2a(eIf2a)、肿瘤抑制蛋白p53、三磷酸肌醇受体(IP3R)、AMPK等;负责自噬诱导的细胞内信号通路主要包括分别在Toll样受体(TLR)4和TLR9激活下的5腺苷单磷酸酯活化蛋白激酶和P38丝裂原活化蛋白激酶(MAPK)通路[5],另外,各种类型的ATG与自噬密切相关,其中Atg7对自噬体的形成有巨大贡献,据有关报道显示其对免疫稳态有益,在CLP手术后8小时表达增强,24小时的时候明显抑制,与自噬液泡数量随时间推移变化的趋势一样[6-7]。Atg1的同源物ULK1和ULK2在TORC1的下游发挥作用,与Atg13、Atg101复合物结合局灶性粘附激酶家族相互作用成FIP200,是哺乳动物自噬体形成的必要条件。在整个自噬的过程中,囊泡的形成较为复杂,需要由Vps34、Vps15、Atg14和Beclin1组成的Ⅲ类磷脂酰肌醇3-激酶复合物来完成噬菌体膜的初始成核和拼装。随后自噬体延伸,此过程中Atg7、Atg10、Atg5、Atg16 I经过一系列结合重组形成Atg12-5-16 I复合物;LC3-PE 由LC3-I与脂质磷脂酰乙醇胺在Atg3和Atg7的作用下结合形成,其在自噬体的延伸中扮演着重要的角色,当细胞延伸到一定程度,包裹着即将被降解的物质与溶酶体融合,随后进行降解,并释放产物,在这个过程中,Atg22发挥自噬降解氨基酸的外排作用。降解过程中若清除破坏细胞中的正常成分则会引起细胞凋亡,影响机体的正常细胞稳态环境,所以有效调控自噬与凋亡的平衡也至关重要。
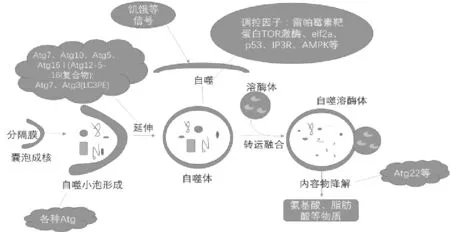
图1 自噬过程简图
2 脓毒症免疫细胞自噬
由某些细菌、细菌毒素(如脂多糖和促炎细胞因子)引起的脓毒症在发生后开始自噬,并被整合到免疫系统的各种功能和过程中。它是机体的重要防御环节,在诱导和调节先天免疫细胞炎症反应中起着至关重要的作用,这些炎症反应是影响脓毒症进展的重要因素。有研究表明,自噬过程和独立的自噬相关蛋白都参与清除病原体、抗原呈递和维持免疫稳态等[8-9]。
2.1 清除细胞内的病原微生物
自噬能够以选择性自噬的形式降解清除细胞内病原体,包括细菌、病毒和真菌等,这种形式被称为异体吞噬,尤其是巨噬细胞自噬被认为是宿主免疫防御的重要组成部分[10]。自噬缺失的小鼠对细菌感染更敏感,对侵犯的病原体和受损细胞器的革除会受到干扰。病原体通过PAMPs、毒素和Ⅲ型或Ⅳ型细菌分泌系统分泌的效应蛋白等诱导自噬的发生,在被感染的巨噬细胞中诱导自噬可以促进包括S. typhimurium和S.flexneri等在内的病原菌的清除。自噬借助Galectin-8信号识别细菌,消除某些从吞噬体逃离的细菌,对于一些例如幽门螺旋杆菌等感染后无法避开吞噬体的细菌,则该包含细菌的吞噬体被自噬体隔离或者与自噬体融合形成双膜或多膜室。除了通过典型的自噬将自噬体中的细菌隔离或通过LAP将自噬体中的细菌隔离外,自噬还可利用P62蛋白将泛素化的胞质蛋白传递到自噬溶酶体,转化为新的杀菌肽有效杀死M.tuberculosis。
脓毒症继发性感染与清除入侵病原体和释放促炎细胞因子的能力有关,中性粒细胞属于机体的第一级防御系统,对病原体的清除至关重要,然而中性粒细胞迁移紊乱或迁徙失当至感染部位,被称为中性粒细胞麻痹,常表现为细菌负荷失控和多器官功能障碍,是致命脓毒症的一大威胁。自噬可以增强免疫缺陷患者中性粒细胞的杀菌活性,杀死入侵的细胞内细菌、释放氧气,有益于脓毒症患者[11],此外,自噬能够调节中性粒细胞的趋化和充盈,降低中性粒细胞充血,对脓毒症多器官功能障碍患者有一定帮助作用,提高患者生存率[5]。
单核细胞和巨噬细胞亦可以清除入侵的病原体和受损组织,有研究表明,单核细胞和巨噬细胞的功能障碍是脓毒症继发性感染增加的主要原因, Lee S等人通过使用Beclin-1杂合敲除小鼠,探讨自噬在脓毒症中的作用,发现Becnl+/-小鼠比同窝出生的野生型小鼠Becnl+/+更易感染,经过CO诱导自噬能够增强巨噬细胞的吞噬能力,降低血液和器官中的细菌浓度,提高脓毒症小鼠存活时间[12]。可见,中性粒细胞、单核细胞和巨噬细胞在脓毒症自噬过程中清除细胞内的病原微生物发挥着重要作用。
2.2 促进抗原呈递
抗原呈递细胞APCs是免疫系统的关键调节因子,抗原经其处理之后由主要组织相容性复合体MHC或相关蛋白将产生的片段呈递给抗原特异性T淋巴细胞,MHC包括MHC-Ⅰ和MHC-Ⅱ类分子,MHC-Ⅰ涉及处理通路主要针对大多数细胞类型的细胞质、细胞核和线粒体中的内源性抗原,而MHC-Ⅱ分子在适当的刺激后,主要呈递外源性抗原和膜性蛋白的抗原多肽。这一现象主要体现在蛋白的两种细胞分解途径:一种是蛋白酶体降解,尤其与MHC-I多肽的产生有关,另一种是核内质溶酶体系统的降解,主要负责MHC-II肽的加工。
自噬分为巨自噬、分子伴侣介导的自噬和微自噬,在真核细胞中广泛存在,在感染过程中自噬介导的抗原呈递对CD4+T细胞的启动活化有重要作用。3-磷酸甘油醛脱氢酶被认为是常见胞质来源的MHC-Ⅱ类的分子配体,在自噬发生后仍能存在自噬体内,且在MHC-Ⅰ类中尚未发现,表明自噬底物由MHC-Ⅱ类分子呈递。对B细胞内的MHC-Ⅱ分子洗脱液中的多肽分析,显示约20%的天然MHC-Ⅱ配体来源于胞质和核蛋白,包括细胞骨架蛋白、热休克蛋白和组蛋白,且在自噬被诱导时,核内抗原呈递增加了50%,膜结合抗原呈递不受影响[13],提示我们内源性蛋白也可以通过MHC-Ⅱ分子表达,自噬可以增强自身抗原的呈递。树突细胞在抗原呈递和连接先天性免疫与适应性免疫系统方面有较强的能力,在感染或损伤刺激下,具有吞噬特性的未成熟树突细胞DC转变为具有抗原呈递作用的成熟状态,从而启动活化T细胞,在脓毒症环境中,DCs在数量和功能上出现异常,扰乱了抗原呈递,有关报道证实自噬能通过增加MHC-Ⅱ分子的表达增强DC抗原呈递功能[14],提示自噬可以作为治疗脓毒症的一种途径。
2.3 维持淋巴细胞稳态
T细胞是适应性免疫的关键因素,越来越多的证据表明自噬对T细胞有保护作用,例如T细胞-特异性Atg7敲除脓毒症小鼠的T细胞大量减少,死亡率升高[6]。在脓毒症模型和临床患者中,细胞凋亡导致T细胞衰竭,脓毒症死亡患者中CD4+和CD8+T细胞显著减少,Takehiko Oami等人试验表明在脓毒症小鼠模型中CD4+T细胞自噬过程不充分、阻断自噬,细胞凋亡增加,死亡率升高[15],而细胞凋亡和自噬有着共同的组成部分,相互调节和修饰各自的活性,并可由饥饿、氧化应激和细胞因子等共同刺激产生,例如共同的有调控蛋白Bcl-2家族和基因Beclin1等,提示自噬和凋亡的协同调控是维持淋巴细胞稳态的关键。
就目前研究来看,自噬调节细胞凋亡的三个过程有:(1)通过特异性自噬蛋白调控细胞凋亡;(2)自噬体膜上Caspase的活化;(3)自噬体的形成和溶酶体活性,细胞凋亡可以通过特异性凋亡蛋白直接调控自噬,也可以利用活化的Caspase对自噬进行调控。自噬的抑制导致细胞对凋亡刺激的敏感性增加,而自噬的增加通过抑制细胞凋亡提高细胞的存活率。有关报道称自噬与调节性T细胞(Treg)的存活和稳定性有关,Treg-特异性基因敲除Atg7和Atg5之后,Foxp3细胞凋亡明显,表达下调,其与自噬耗竭下TCR-mTOR复合体信号过度激活有关,而抑制mTOR也导致Treg显著缺失,此外,Beclin-1和Atg14胞内结构域结合的Treg自噬需要Notch1通路进一步调节细胞存活[10]。B淋巴细胞与T淋巴细胞类似,在发育过程中需要自噬,Atg5缺乏的pro-B细胞不能有效的发育成pre-B细胞,会随着凋亡的增加而死亡。有研究表明[11],HMGB1降低增加自噬,氧化后的HMGB1增加细胞凋亡。由此可见,自噬和凋亡之间的平衡可能影响脓毒症进展过程中的细胞稳态。
3 自噬对脓毒症器官的影响
脓毒症是宿主对自身组织和器官感染损伤的反应,常表现为多器官衰竭。现有多项研究证实自噬对脓毒症中的多个器官具有保护作用,涉及心、肝、肾等。
3.1 对肝的影响
脓毒症或脂多糖诱导的自噬无论在体内还是体外都可以保护肝细胞死亡。Carchman EH等人发现无论是C57BL/6小鼠结扎和CLP后的肝脏,还是LPS(100 ng/mL)处理后的原代小鼠肝细胞,血红素氧合酶HO-1和自噬均上调,使用锡-原卟啉或下调HO-1阻止模型中的自噬,会导致肝细胞损伤、凋亡和死亡的增加[16]。自噬缺陷增加TNF诱导细胞凋亡、ATP耗竭和白蛋白生成减少[17]。京尼平能够增加自噬通量,有助于提高脓毒症小鼠的存活率[18]。有研究显示肝细胞分解产物一方面可以维持代谢需要另一方面又可以限制自噬程度,氨基酸较糖类和脂质调控效果更明显,当氨基酸达到一定量就会激活mTOR信号从而减弱肝细胞的自噬[19],当超出一定范围之后,抑制自噬能力过强,也会减小肝细胞对病理损伤的抵抗能力,加重病情[20]。此外,脓毒症导致的肝损伤中HIPK2表达降低,Zhengyu Jiang等人通过实验发现,HIPK2过表达降低了脓毒症诱导的肝损伤小鼠血清中的炎症细胞因子浓度和相关氧化应激的指标,提高了存活率,改善了肝损伤,在这一过程中,自噬通量增加,表明HIPK2过表达可以通过恢复自噬来改善脓毒症所致的肝损伤[21]。
3.2 对心的影响
心脏是脊椎动物身体中最重要的器官之一,脓毒症引起严重的心肌抑制,超声心动图常显示严重的双心室功能障碍,严重影响人类生活。自噬损伤可能导致心肌细胞收缩功能障碍和凋亡细胞死亡,雷帕霉素诱导的自噬,可以修复CLP诱导的心脏功能障碍,恢复左心室射血分数,保护心肌细胞免于凋亡和坏死,在脓毒症中达到保护心脏的作用。脓毒症时,诱导型一氧化氮合酶2活性升高,产生大量NO,这是自噬的调控因子之一,过量的NO会抑制S-亚硝基化、JNK1和IKKβ活性,NO对JNK1的抑制降低了Bcl-2磷酸化,增加了Bcl-2-Beclin1的相互作用,干扰hVps34/Beclin 1复合体的形成等抑制自噬,影响受损的心肌功能恢复。能量消耗是脓毒症患者心肌功能障碍的主要原因,mTOR是能量状态的主传感器,在能量消耗时促进自噬,脓毒症大鼠mTOR通路受阻,使用雷帕霉素后改善了mTOR通路促进自噬,脓毒症导致的症状有所缓解[22]。
3.3 对脾的影响
脓毒症是对感染的系统性反应,可导致脾损伤。研究发现,脓毒症诱导的淋巴细胞和树突细胞凋亡导致免疫抑制,从而导致无法根除原发性感染及获得新的继发性感染趋势,在脓毒症晚期,大量淋巴细胞发生凋亡现象,患者免疫功能下降,威胁到他们的生命健康。Zhang L等人证明了干扰素调节因子1(IRF-1)在脓毒症模型小鼠中同时调节免疫细胞凋亡和自噬,IRF-1在脾细胞中是通过Toll样受体4、髓样分化初级应答基因88被激活的,IRF-1敲除的小鼠可以通过减少脾细胞凋亡、增加自噬来防止脓毒症害[23]。
3.4 对肺的影响
当前的研究表明脓毒症急性肺损伤和急性呼吸窘迫综合症是脓毒症患者死亡的主要原因之一,根据报道,脓毒症肺损伤患者的死亡率高于40%[24]。自噬可以降解老化和有缺陷的细胞器,但当自噬通量被破坏时,有害的蛋白质或细胞器例如线粒体会累积,并将进一步损伤肺组织。PICK1在许多组织中含量丰富,特别是在大脑和睾丸中,并在肺中适当表达,通过抑制巨噬细胞极化,证实其在脂多糖诱导的急性肝损伤中具有抗炎作用,Yunchang Mo等人通过建立体内和体外脓毒症模型,探索了PICK1的功能及其与自噬的关系,发现PICK1的缺失会导致自噬通量的破坏,并加重脓毒症引起的肺损伤[25]。LC3转基因可能通过增加自噬体清除率来减轻脓毒症中的肺损伤和炎症。然而,由于自噬在血管平滑肌细胞中具有VSMCs双面表型转化,因此在减轻肺损伤时需要维持在适当得水平[23]。
3.5 对肾的影响
肾是人体最重要的器官之一,脓毒症引起肾损伤甚至肾功能衰竭,严重危害人们的生活水平和健康,有报道称自噬能够保护肾小管上皮细胞,对肾脏损伤的恢复有益[26]。向镜芬等人发现,在CLP肾脏损伤后,使用PI3K和Akt抑制剂后,自噬相关分子蛋白表达量增加,HK-2细胞凋亡增加,LC3等自噬相关蛋白减少,表明脓毒症肾损伤中的自噬与PI3K/Akt通路有关[27],可以作为治疗的一种途径。此外,Atg7的下调会增加脓毒症引起的急性肾功能障碍,3-甲基腺嘌呤对自噬的抑制作用显著提高肾小管的损伤。由此可见自噬在脓毒症造成的肾功能障碍恢复中发挥着重要的作用。
4 结语
脓毒症的发病人群涉及较广,低至新生儿,高至老年人,与免疫系统密切相关,其发生发展严重威胁人类的生命健康,给人们的生活带来了极大的困扰,面对这个发病率高、死亡率高的疾病,寻找其有效的治疗方法势在必行。自噬能够适应性的保护机体,降解有害、多余的细胞成分,清除有害的病原体,维持细胞环境稳态,自噬无论在正常生理状态还是病理情况下都是维持机体稳态的关键。多年的研究表明,自噬可以作为一种新型的治疗方法来治疗癌症、神经退行性病变等疾病。自噬可以被认为是对感染的先天免疫反应,不仅对炎性小体激活有抑制作用,而且对不依赖于CASP1激活的炎症介质也有抑制作用,除了对促炎细胞因子的影响,还对TLR信号通路有影响,能够改善微生物配体向促进I型IFN合成的TLR7等细胞质受体的传递,在宿主防御过程中自噬和炎症相互交织,对多种疾病的发病机制和治疗有重要影响[28]。脓毒症急性期有强烈的炎症反应,晚期免疫系统向抗炎、免疫抑制状态转换,炎症减轻,开始组织修复,现代治疗方法有LUCI、CRRT、HFNC等,但死亡率仍然高达20%~25%,以往的研究中,许多免疫调节方法虽已在脓毒症临床试验中进行了测试,但仍没得出安全有效的治疗方法[29],脓毒症体内外模型显示,自噬在脓毒症中被激活,不仅利于免疫细胞的存活,对维持宿主免疫系统在脓毒症环境下的功能稳态也有益处,且随脓毒症的发展,自噬动力学也会相应的发生变化,且涉及多个免疫细胞受体和细胞内信号通路,对脓毒症造成的器官功能障碍有恢复和保护作用,可以作为一种治疗脓毒症的途径,但值得注意的是在疾病的炎症过程没有得到适当控制的时候,自噬可能会产生负面作用,对宿主有害。所以结合自噬的机制特点与凋亡发生条件等综合因素,有效平衡和控制自噬的程度,从而达到治疗脓毒症、提高脓毒症患者生存率的目的的研究是非常有必要的。此外,大多研究表明[30],自噬在疾病晚期受到抑制,提示在脓毒症的某个恰当阶段对自噬进行调控可能也是一种治疗脓毒症的途径。
猜你喜欢
大电机技术(2022年3期)2022-08-06
核科学与工程(2021年4期)2022-01-12
新世纪智能(数学备考)(2021年10期)2021-12-21
煤气与热力(2021年4期)2021-06-09
中华养生保健(2020年4期)2020-11-16
中华戏曲(2020年1期)2020-02-12
中国中医急症(2019年10期)2019-05-21
中成药(2017年12期)2018-01-19
中国交通信息化(2017年8期)2017-06-06
中华老年多器官疾病杂志(2016年9期)2016-04-28
