
气相色谱法测定亚甲基二膦酸原料药中两种工艺杂质的含量
2019-04-08 07:17朱志玲张蕴涵山广志
同位素 2019年2期
王 冉,朱志玲,张蕴涵,胡 骥,姚 静,山广志
(1.中国医学科学院 医药生物技术研究所,北京 100050;2.原子高科股份有限公司,北京 102413;3.中国食品药品检定研究院,北京 100050)
锝[99Tc]亚甲基二膦酸盐注射液(云克)在临床上主要用于类风湿性关节炎治疗,具有抗炎、镇痛和免疫调节及破骨修复作用[1-4]。锝[99mTc]亚甲基二膦酸盐注射液作为骨显像剂,用于早期诊断恶性转移性骨肿瘤和原发性骨肿瘤,单次注射使用。
亚甲基二膦酸(methylenediphosphonic acid, MDP)作为锝[99Tc]亚甲基二膦酸盐注射液和锝[99mTc]亚甲基二膦酸盐注射液的主要原料,多采用温和的亚磷酸三异丙酯合成路线[5],在其合成工艺中可能存在两种工艺杂质(原料及中间体):亚磷酸三异丙酯(triisopropyl phosphite, TPP)和亚甲基二磷酸四异丙酯(tetraisopropyl methylenediphosphonate, TPMDP)。
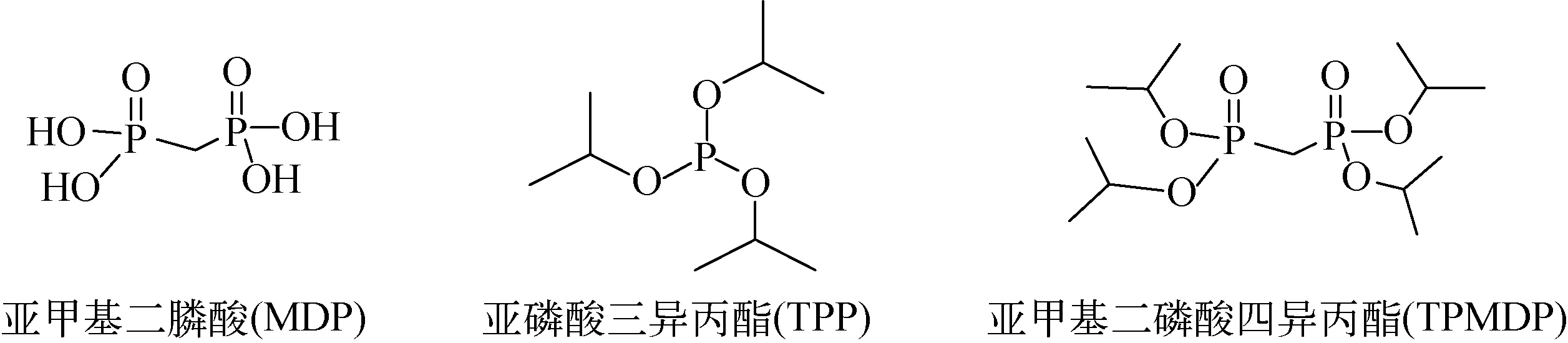
图1 MDP及两种杂质结构Fig.1 Structures of MDP and two impurities
中国药典收录了锝[99mTc]亚甲基二膦酸盐注射液的质量标准[6],但未收载MDP原料药的质量标准,检索CFDA网站,亦未发现MDP原料药注册信息。欧洲药典[7]采用标准加入法,利用核磁共振氢谱法(1H NMR)对MDP原料药中可能残留的TPP和TPMDP进行控制,并规定两种工艺杂质残留量均不得高于1%。
欧洲药典收载的NMR方法需要使用昂贵的专用设备,无法满足生产控制和一般实验室日常检测的需求。本实验采用气相色谱法[8-11]建立MDP原料药中两种工艺杂质的气相色谱-氢火焰离子化检测器(GC-FID)控制方法,为MDP原料药中两种工艺杂质的检测和控制提供了一种简便可行的分析方法,为MDP原料药的质量控制提供了方法和依据,能够有效保证以MDP为原料药的临床药品的质量安全。
1 仪器与试剂
TRACE1310气相色谱仪:美国Thermo Fisher公司;ME204型电子天平、XP205型电子天平:瑞士Mettler-oledo公司;Milli-Q Integral超纯水器:德国Merck Millipore公司。
TPP:纯度97.88%,北京百灵威科技有限公司,批号L360R28;TPMDP:纯度99.40%,北京百灵威科技有限公司,批号LPB0058;MDP:原子高科股份有限公司提供,批号10201922、10208056、10032224。水为超纯水,其他试剂为分析纯。
2 方法与结果
2.1 色谱条件
以硝基对苯二甲酸改性的聚乙二醇为固定液的毛细管柱(HP-FFAP)(25 m×0.32 mm,0.5 μm)为色谱柱;程序升温,初始温度为40 ℃,保持3 min,以15 ℃/min升温至220 ℃,保持6 min;载气为氮气,流速为2.5 mL/min;进样口温度为200 ℃;不分流进样,进样量1.0 μL。
2.2 溶液的制备
2.2.1对照品储备液 精密称取TPP和TPMDP对照品适量,用丙酮稀释至浓度为400 mg/L,摇匀,作为对照品储备液。
2.2.2对照品溶液 精密量取上述对照品适量,用丙酮稀释至100 mg/L,即得。
2.2.3供试品溶液 取MDP(批号:10201922)约20 mg,精密称定置20 mL顶空瓶中,精密加入丙酮2 mL,密封,涡旋震荡6 min,静置,取上清,作为供试品溶液。
2.2.4供试品加标溶液 取MDP(批号:10201922)约20 mg,精密称定置20 mL顶空瓶中,精密加入对照品溶液2 mL,密封,涡旋震荡6 min,静置,取上清,作为供试品加标溶液。
2.3 专属性
取丙酮、对照品溶液、供试品溶液、供试品加标溶液各1 μL,分别注入气相色谱仪,按“2.1”小节色谱条件测定,记录色谱图,结果示于图2,TPP保留时间为5.947 min,TPMDP保留时间为15.547 min,空白溶剂丙酮和供试品中其他色谱峰均不干扰目标峰测定。
2.4 溶液稳定性
取供试品加标溶液,分别于0、1、2、4、8 h进样,记录TPP和TPMDP峰面积(图3)。结果8 h内TPP峰面积RSD=12.57%,TPMDP峰面积RSD=3.15%。室温条件下,TPMDP 8 h内保持稳定,TPP在供试品溶液中只能在2 h内保持稳定。因此供试品溶液采用临用现制的方法进行制备。
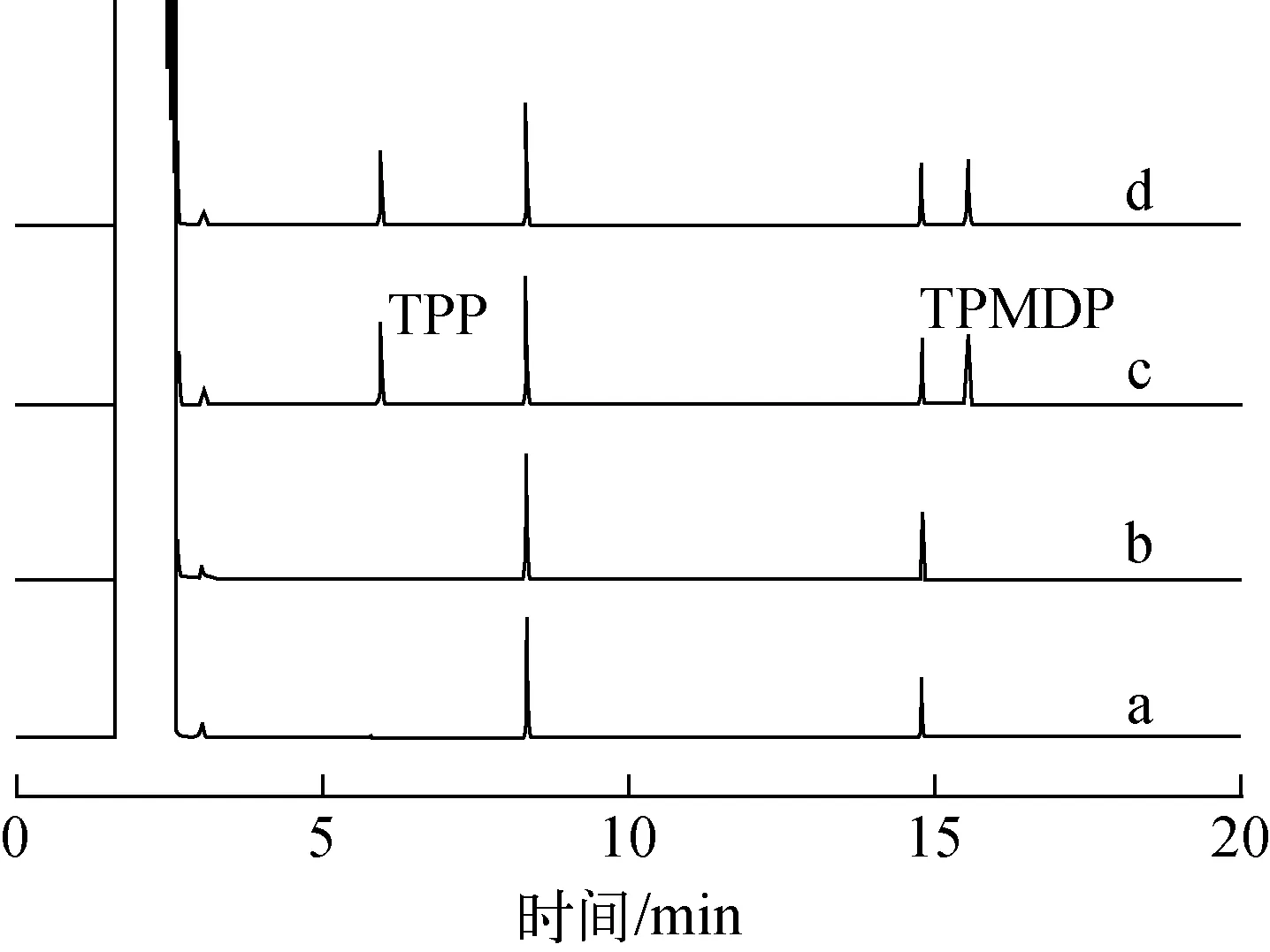
a——空白溶液;b——供试品溶液;c——对照品溶液;d——供试品加标溶液;图2 MDP中两种杂质色谱图a——Blank solution;b——Test sample;c——Reference solution;d——Test spiked solution;Fig.2 GC chromatograms of two impurities of MDP
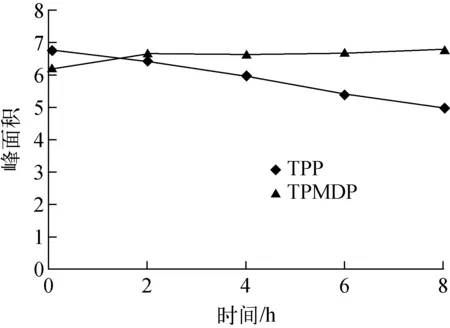
图3 供试品加标溶液中TPP和TPMDP 8 h内峰面积变化图Fig.3 Change of peak areas within 8 hours of TPP and TPMDP in the spiked solution
2.5 线性关系
精密量取“2.2.1”小节对照品储备液,用丙酮依次稀释成TPP浓度分别为194.59、97.29、48.65、24.32、12.16、6.08 mg/L,TPMDP浓度分别为200.69、100.34、50.17、25.09、12.54、6.27 mg/L的系列对照品溶液,按“2.1”小节色谱条件,进样测定。以峰面积(A)为纵坐标,浓度(C)为横坐标,进行线性回归。TPP在6.081~194.6 mg/L范围内,浓度与峰面积线性关系良好,回归方程为A=0.075C-0.095(r=0.999,n=6);TPMDP在6.272~200.7 mg/L浓度范围内,浓度与峰面积线性关系良好,回归方程为A=0.066C-0.036(r=0.999,n=6)。
2.6 精密度
取对照品溶液,按“2.1”小节色谱条件连续进样6次,结果TPP和TPMDP峰面积RSD分别为2.00%、2.08%,方法精密度良好。
2.7 检出限和定量限
精密量取“2.2.1”小节对照品储备液加丙酮逐级稀释,按“2.1”小节色谱条件进样,并记录色谱图,按信噪比(S/N)分别为10和3计算定量限和检出限。TPP的定量限为1.520 mg/L,检出限为0.760 1 mg/L;测得TPMDP的定量限为1.568 mg/L,检出限为0.783 9 mg/L。可满足两种工艺杂质的测定要求。
2.8 重复性
精密称取6份供试品,按“2.2.4”小节方法制备供试品加标溶液,按“2.1”小节色谱条件,进样测定。TPP测得量的RSD=1.73%,TPMDP测得量的RSD=3.45%,方法重复性良好。
2.9 回收率
取供试品约20 mg,精密称定,置20 mL顶空瓶中,按高、中、低不同浓度精密加入混合对照品溶液适量,每个浓度各3份,按“2.2.4”小节方法制备供试品加标溶液,进样测定,计算回收率。两种杂质的平均加样回收率(n=9)分别为96.04%,97.72%。

表1 TPP回收率结果(n=9)Table 1 The recovery result of TPP (n=9)
ND:未检出
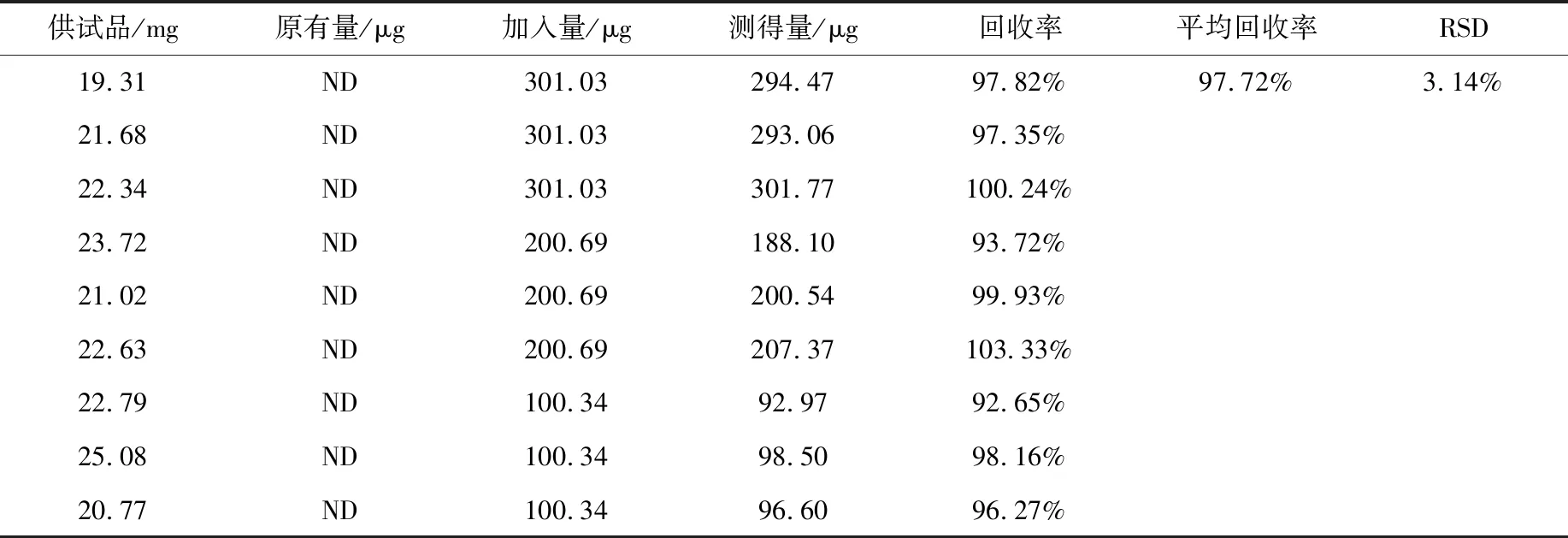
表2 TPMDP回收率结果(n=9)Table 2 The recovery result of TPMDP (n=9)
ND:未检出
2.10 耐用性
载气流速(±2%)、进样口温度(±2 ℃)、检测器温度(±2 ℃)发生微小变化时,TPP测定的含量RSD分别为:3.37%、0.46%、3.05%;TPMDP测定的含量RSD分别为:2.68%、0.89%、1.62%。表明方法耐用性良好。
2.11 样品测定
取本品3批样品,按照“2.2”小节方法制备供试品溶液和对照品溶液,按“2.1”小节色谱条件,进样测定,记录色谱图,结果3批样品中均未检出TPP和TPMDP。
利用Bruker 600 MHZ NMR,按照欧洲药典[7]采用标准加入法对供试品(批号:10201922)和对照溶液中TPP和TPMDP的含量进行测定,样品中均未检出两种工艺杂质,供试品中的杂质含量符合限度要求。1H NMR测定谱图示于图4。

1——TPP定量峰;2——TPMDP定量峰图4 样品核磁测定图谱1——Quantitative peak of TPP;2——Quantification of TPMDPFig.4 Sample’s NMR determination map
3 讨论
3.1 溶剂
分别采用乙酸乙酯、丙酮、三氯甲烷、二氯甲烷溶剂进样实验,其中二氯甲烷、三氯甲烷、丙酮作为溶剂时,均不干扰TPP和TPMDP的测定,乙酸乙酯溶剂峰干扰TPP测定,因此本实验最终选择丙酮作为溶剂。
3.2 提取时间
对提取时间进行考察,在涡旋振荡仪上准确震荡3、6、9、12、15 min,发现在实验过程中震荡时间超过6 min后测定结果几乎无变化,所以选取6 min作为样品的提取时间。
3.3 TPP稳定性
室温条件下,TPP在丙酮溶液中能够长时间保持稳定,精密度良好;而在供试品加标溶液中,TPP仅能在2 h内保持稳定,推测该现象可能是由于TPP在酸性条件下不稳定,发生降解导致。故采用临用现制的方法进行样品制备。
3.4 GC-FID法与NMR方法灵敏度的比较
比较NMR和GC-FID方法对上述工艺杂质的检测灵敏度(图5)。结果显示, NMR方法测定TPP的检出限为70.51 mg/L,TPMDP的检出限为78.75 mg/L。GC-FID方法检测灵敏度较NMR方法更高。

1——TPP定量峰;2——TPMDP定量峰图5 核磁测定MDP两种杂质灵敏度图谱(TPP:70.51 mg/L;TPMDP:78.75 mg/L)1——Quantitative peak of triisopropyl phosphonate;2——Quantification of methylene diphosphonate tetraisopropyl esterFig.5 The sensitivity map of two impurities in MDP measured by nuclear magnetism(TPP:70.51 mg/L;TPMDP:78.75 mg/L)
4 结论
本研究建立了同时测定MDP原料药中两种工艺杂质残留量的气相色谱方法。建立的方法专属性强、灵敏度高,能够有效控制MDP原料中的可能杂质。建立的方法为药品生产企业提供了简便的控制手段,对提高MDP原料药的质量控制水平,保障用药安全具有推动作用,对促进MDP原料药的研发和申报具有一定借鉴价值。
猜你喜欢
中国经济周刊(2021年22期)2021-12-07
昆明医科大学学报(2021年4期)2021-07-23
化工管理(2021年10期)2021-04-25
艺术品鉴(2020年6期)2020-12-06
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
分析化学(2019年3期)2019-03-30
安徽化工(2018年2期)2018-05-22
中国经济信息(2017年17期)2017-09-09
领导文萃(2017年6期)2017-03-24
