
HPLC-MS/MS法同时测定鸡肉中40种糖皮质激素和9种非甾体抗炎药物残留
2019-03-28 09:52陈晶燕陈万勤梁晶晶丁宇琦赵超群罗金文
质谱学报 2019年2期
陈晶燕,陈万勤,刘 柱,梁晶晶,丁宇琦,赵超群,罗金文
(1.浙江工业大学,浙江 杭州 310014;2.浙江省食品药品检验研究院,浙江 杭州 310052)
糖皮质激素是一种固醇类激素,在调节免疫应答反应中起着重要作用[1],具有抗炎、抗过敏和免疫抑制等作用。糖皮质激素还可以促进动物生长,因此有一些饲养者非法过量使用这些药物以促进动物生长、降低饲养成本。然而,残留在动物源性食品中的激素会影响食用者健康,导致高血压、糖尿病、代谢系统紊乱和骨质疏松[2]等疾病。非甾体抗炎药是一类不含有甾体结构的抗炎药,具有抗炎和解热镇痛的作用。目前,非甾体抗炎药在养殖业中应用越来越广泛,但其在动物源性食品中的过量残留会导致食用者发生高血压、心肌梗死、心力衰竭和心律失常[3]等疾病。因此,许多国家都制定了动物源性食品糖皮质激素和非甾体类抗炎药的严格限量标准。我国农业部第235号公告[4]规定:地塞米松和倍他米松在猪、牛的肌肉和肾脏中不得超过0.75 μg/kg,肝脏中不得超过2 μg/kg,牛奶中不得超过0.3 μg/kg;地西泮在所有动物食品中均不得检出。
目前,糖皮质激素和非甾体类抗炎药物的测定方法主要有高效液相色谱法[5-6]、液相色谱-质谱法[7-9]、气相色谱-质谱法[10]以及毛细管电泳法[11]等。其中,液相色谱-串联质谱法因其高灵敏度和高选择性,成为糖皮质激素和非甾体类抗炎药物残留分析中最常用的手段之一。常用的样品前处理方法主要有固相萃取[12]、液-液萃取[13]、免疫亲和色谱[14]和凝胶渗透色谱[15]等,但是存在操作复杂、费时等缺陷,不适用于大量样品的快速检验。
鸡肉样品中基质比较复杂,为减少基质干扰,本工作拟选取90%乙腈-0.1%甲酸混合溶液提取目标化合物,经Oasis PRIME HLB固相萃取柱净化,采用超高效液相色谱-串联质谱法同时测定鸡肉中40种糖皮质激素和9种非甾体抗炎药物残留,希望建立一种适用于日常大批量样品的定量检测方法。
1 实验部分
1.1 仪器与试剂
Agilent 6460 LC-MS/MS液相色谱-质谱仪:美国Agilent公司产品;Milli-Q超纯水器:美国Millipore公司产品;Acquity UPLC BEH C18色谱柱(2.1 mm×100 mm×1.7 μm),Waters Oasis PRIME HLB固相萃取柱(6 mL, 200 mg):美国Waters公司产品。
甲醇、乙腈:均为质谱纯,德国Merck公司产品;甲酸:质谱纯,美国Sigma Aldrich公司产品;其它化学试剂均为分析纯。鸡肉为市场购买。
40种糖皮质激素药物:曲安西龙、氢化可的松、倍他米松、地塞米松、氟米松、倍氯米松、氢化可的松醋酸酯、地夫可特、甲基泼尼松龙醋酸酯、曲安奈德醋酸酯、倍他米松戊酸酯和安西奈德,中国食品药品检定研究院产品;泼尼松龙和泼尼松,中国药品生物制品检定研究所产品;可的松、氟氢可的松醋酸酯和泼尼松醋酸酯,加拿大TRC公司产品;甲基泼尼松龙、氟氢缩松、曲安西龙双醋酸酯、泼尼松龙醋酸酯、氟米龙、倍他米松醋酸酯、布地奈德、氢化可的松丁酸酯、地塞米松醋酸酯、氟米龙醋酸酯、氢化可的松戊酸酯、氟轻松醋酸酯、二氟拉松双醋酸酯、泼尼卡酯、阿氯米松双丙酸酯、莫米他松糠酸酯和倍氯米松双丙酸酯,美国药典委员会产品;曲安奈德、可的松醋酸酯、氯倍他索丙酸酯和倍他米松双丙酸酯,德国Dr. Ehrenstorfer公司产品;氟替卡松丙酸酯和氯倍他松丁酸酯,欧洲药品质量管理局产品。
9种非甾体抗炎药物:美洛昔康、醋氯芬酸、地西泮和氯苯那敏,中国食品药品检定研究院产品;吡罗昔康、保泰松和罗通定,中国药品生物制品检定研究所产品;依托考昔,加拿大TRC公司产品;塞来昔布,欧洲药品质量管理局产品。
49种药物的纯度均大于97%。
1.2 标准溶液的配制
49种药物标准储备溶液:分别准确称取各约10 mg的40种糖皮质激素和9种非甾体抗炎药物于25.0 mL容量瓶中,用甲醇溶解并定容,配制成约0.4 g/L的标准储备液,于-18 ℃避光保存,备用。
49种药物混合标准储备溶液:分别准确量取适量各单一成分对照品标准储备溶液,混合后用30%乙腈-水溶液稀释,配制成1 mg/L的混合标准储备液,于4 ℃避光保存,备用。
1.3 样品前处理
1.3.1样品提取 称取2.00 g均匀制备的鸡胸肉泥样品于50 mL高速离心管中,加入10 mL 90%乙腈(含0.1%甲酸溶液)摇匀,采用高速均质器以15 000 r/min均质约2.0 min,超声提取10 min,以5 000 r/min离心5 min,取上层清液进行下一步净化处理。
1.3.2样品净化 准确移取5 mL 1.3.1节得到的样品上清液,过Oasis prime HLB固相萃取柱,控制流速1~2滴每秒,收集流出液,待样液全部通过固相萃取柱后,用4 mL 90%乙腈-0.1%甲酸溶液清洗,收集全部洗脱液,40 ℃下氮气吹至液面低于1 mL,并用30%乙腈溶液转移定容至1.0 mL,漩涡混匀,过0.22 μm有机微孔滤膜,转移至样品瓶中,供LC-MS/MS分析。
1.4 LC-MS/MS条件
1.4.1色谱条件 Waters Acquity UPLC BEH色谱柱(2.1 mm×100 mm×1.7 μm);流动相:A为0.1%甲酸溶液,B为乙腈;梯度洗脱程序:0~3 min(30%B),3~12 min(30%~75%B),12~14 min(75%),14~14.01 min(75%~30%B);14.01~17 min(30%B);流速0.3 mL/min;柱温35 ℃;进样量5 μL。
1.4.2质谱条件 采用电喷雾离子源(Jet stream ESI),49种药物均采用正离子模式检测;多反应监测模式(MRM);电离电压4.0 kV(正离子扫描),-3.5 kV(负离子扫描);干燥气流速7 L/min,温度300 ℃;鞘气流速10 L/min,温度350 ℃;雾化气流量2.07×105Pa。49种药物的监测离子对及相关参数设定列于表1。
2 结果与讨论
2.1 提取溶液的优化
糖皮质激素类药物属于弱极性化合物,易溶于有机溶剂,难溶于水。非甾体抗炎药属于酸性或中性药物,在酸性条件下更易溶于有机溶剂。实验考察了乙腈、甲醇和乙酸乙酯作为提取溶剂的提取效果。结果表明,甲醇作为提取溶剂对糖皮质激素的提取效果较差;乙酸乙酯对糖皮质激素类药物的提取效果较好,但是对非甾体抗炎药的提取效果较差,且提取液中脂肪较多,不利于后续固相萃取净化。乙腈对两类药物的提取效率优于甲醇和乙酸乙酯,对糖皮质激素的提取效率高于非甾体抗炎类药物。

表1 49种药物的质谱采集参数Table 1 Mass spectrometry parameters of 49 veterinary drugs
注:*为定量离子
为进一步提高提取液的提取效率,实验比较了含不同浓度甲酸提取液的提取效果。考察了乙腈中加入0.1%、0.2%、0.3%甲酸以及不加甲酸的乙腈对目标物的提取效率。结果表明,加入甲酸后,各药物的回收率都有所提高,其中吡罗昔康、美洛昔康、醋氯芬酸和保泰松的回收率明显提高,且含0.2%、0.3%甲酸提取试剂的回收率与含0.1%甲酸提取试剂的回收率并无显著的组间差异。因此,本实验选择含0.1%甲酸的乙腈作为提取液。
另外,实验还比较了不同比例的乙腈水溶液的提取效果。分别采用乙腈含量为70%、80%、90%、95%的溶液(均含有0.1%甲酸溶液)进行提取效率优化实验。结果表明,90%、95%和100%乙腈的提取率相对较高。此外,采用90%乙腈作为提取溶剂时,回收率较稳定,其原因可能是提取液含10%水,可以有效地降低提取液中的脂肪含量,从而减少净化过程中脂肪的影响,因此选择0.1%甲酸-90%乙腈溶液作为提取剂。
2.2 净化条件的优化
鸡肉中的基质较为复杂,包括蛋白质、脂肪、矿物质和磷脂等物质,提取后需净化才能进行质谱分析,否则不仅会产生很严重的基质效应,还会污染质谱检测器。样品中的蛋白质在
90%乙腈水溶液中会发生变性,可通过高速冷冻离心沉淀去除,在高速冷冻离心的过程中,还可同时去除一部分脂肪和磷脂;然后,实验采用Waters Oasis PRIME HLB、Waters Oasis HLB、Waters Sep-Pak C18三种固相萃取柱进行样品净化。其中,Oasis HLB和C18小柱要求在大比例水相的条件下上样,故需先将样品稀释再上样。实验结果表明,Oasis HLB和C18小柱无法保留全部药物,使用Oasis HLB固相萃取柱时,氯苯那敏、美洛昔康和醋氯芬酸的回收率较低;使用C18小柱时,氯苯那敏、罗通定、吡罗昔康和美洛昔康的回收率较低。Oasis Prime HLB固相萃取柱采用一步净化,通过保留样品基质中的杂质,流出目标物的方式实现样品溶液的净化,简化了操作步骤,既有效除去了磷脂、脂肪等杂质干扰,又减小了待测组分损失。实验对比了经过Oasis Prime HLB固相萃取柱净化前后,磷脂类物质的净化效果。采用磷脂类物质特征子离子m/z184,扫描其母离子的方式进行检测,结果示于图1。净化前样品的质谱响应约为7×106,净化后样品的质谱响应约为0.5×105,净化率达到99%。综上所述,本实验选择Oasis PRIME HLB固相萃取柱对样品进行净化。
2.3 液相色谱条件的优化
40种糖皮质激素类和9种非甾体抗炎药物在C18色谱柱上均有较强的保留。本方法在相同的流动相比例下分别考察了几种C18色谱柱对49种药物残留的分离效能,发现Waters Acquity UPLC BEH C18色谱柱(2.1 mm×100 mm×1.7 μm)的分离效果较好。

图1 样品提取液经Oasis PRIME HLB净化前(a)、后(b)的总离子流图Fig.1 Total ion chromatograms of the sample before (a) and after (b) purified by Oasis PRIME HLB
糖皮质激素类药物的化学结构相似,存在多个同分异构体,因此在分离上存在一定难度。采用甲醇体系流动相时,各化合物无法完全分离,并且杂质干扰较多,目标化合物响应值较低。乙腈为流动相不仅能使同分异构体分离,响应值较高,而且还可以得到较完美的峰形。甲酸是糖皮质激素电离诱导过程不可缺少的物质,而且非甾体抗炎药在酸性条件下更易溶于乙腈。实验对比了乙腈与水、乙酸铵溶液、甲酸水溶液等多种流动相。结果表明,以乙腈与水为流动相会造成峰拖尾;以乙腈与乙酸铵水溶液为流动相会造成部分药物峰形较差;以乙腈与0.1%甲酸水溶液为流动相时的分离效果最好且目标离子的信号响应值较高。因此,确定流动相为乙腈与0.1%甲酸水溶液。
2.4 质谱条件的优化
由于大部分非甾体抗炎药含有电负性基团,在正离子模式下易结合质子形成准分子离子[M+H]+。糖皮质激素在正离子模式下得到的碎片离子信息多于负离子模式。在正离子扫描模式下,用Optimizer软件对1.0 mg/L的所有化合物的单标溶液分别进行质谱参数优化,找到主要碎片离子、最优碰撞电压和碎裂电压,选择干扰小、灵敏度高的离子作为定性离子,每种化合物选择两对离子,满足欧盟委员会指令96/23/EC对定性分析的要求,并依据子离子相对丰度比(定性离子/定量离子)和化合物保留时间进行定性。利用MRM模式在优化的液相色谱和质谱条件下可以有效分离49种目标化合物,各药物MRM色谱图示于图2。由于化合物较多,将保留时间相近的化合物置于不同的提取离子流图中。
49种化合物中共有6组同分异构体,分别为泼尼松龙(2)和可的松(5),泼尼松龙醋酸酯(14)和可的松醋酸酯(20),氟轻松醋酸酯(29)和二氟拉松双醋酸酯(30),倍他米松(7)和地塞米松(8),曲安奈德(11)、倍他米松醋酸酯(22)和地塞米松醋酸酯(25),阿氯米松双丙酸酯(33)和倍氯米松双丙酸酯(39)。其中,前三组同分异构体的定量子离子和定性子离子不同,可根据提取定性离子对得到不同的色谱图判定化合物。后三组同分异构体的定性/定量离子相同,如化合物7、8的定性/定量离子对均为m/z393.2/146.8,难以根据碎片离子进行区别,但二者的保留时间分别为4.48、4.68 min,可根据其保留时间区别化合物7和8。
2.5 方法学考察
2.5.1线性范围、检出限和定量限 采用外标法定量,为了尽可能消除基质效应带来的测量误差,采用空白样品提取液作为标准溶液的配制溶剂。称取约2 g不含目标物的样品于50 mL离心管中,按1.3节方法进行前处理,得到样品基质溶液。精密量取一定体积1 mg/L的混合标准储备溶液,用样品基质溶液稀释,配制成一系列浓度分别为2、5、10、50、100 μg/L的基质标准曲线工作溶液,在最佳的仪器条件下对标准溶液进行检测,以定量离子峰面积对质量浓度进行线性回归。结果表明,在2~100 μg/L范围内,49种药物的线性关系良好,线性相关系数均大于0.996。
采用基质液添加各药物标准溶液,分别以3倍和10倍信噪比(S/N)确定检出限和定量限,结合各种药物的残留限量标准,确定曲安西龙、泼尼松、可的松、甲基泼尼松龙、氟氢缩松、泼尼松龙醋酸酯、甲基泼尼松龙醋酸酯、莫米他松糠酸酯、保泰松、地西泮的检出限为0.5 μg/kg,定量限为1.5 μg/kg;泼尼松龙、倍氯米松、曲安西龙双醋酸酯、可的松醋酸酯、倍他米松醋酸酯、布地奈德、氢化可的松丁酸酯、地塞米松醋酸酯、倍氯米松双丙酸酯、氯倍他松丁酸酯、美洛昔康、吡罗昔康、塞来昔布的检出限为0.3 μg/kg,定量限为1.0 μg/kg;其余化合物的检出限为0.1 μg/kg,定量限为0.3 μg/kg。
2.5.2加样回收率实验和精密度考察 对不含目标物的空白样品进行加标回收率实验,添加水平分别为2、10和20 μg/kg,采用优化后的方法进行前处理,每个浓度水平进行6次平行测试,计算回收率及相对标准偏差(RSD)。2 μg/kg加标水平下,49种药物的回收率为64.5%~111.7%,RSD为3.5%~12.0%;10 μg/kg加标水平下的回收率为60.2%~100.4%,RSD为3.4%~11.9%;20 μg/kg加标水平下的回收率为61.6%~98.5%,RSD为1.3%~10.3%。以上结果表明,该方法的准确度和精密度良好,能够满足鸡肉中糖皮质激素和非甾体抗炎药测定的要求。
因篇幅所限,详细实验数据以附件形式提供,请登录本刊网站下载。
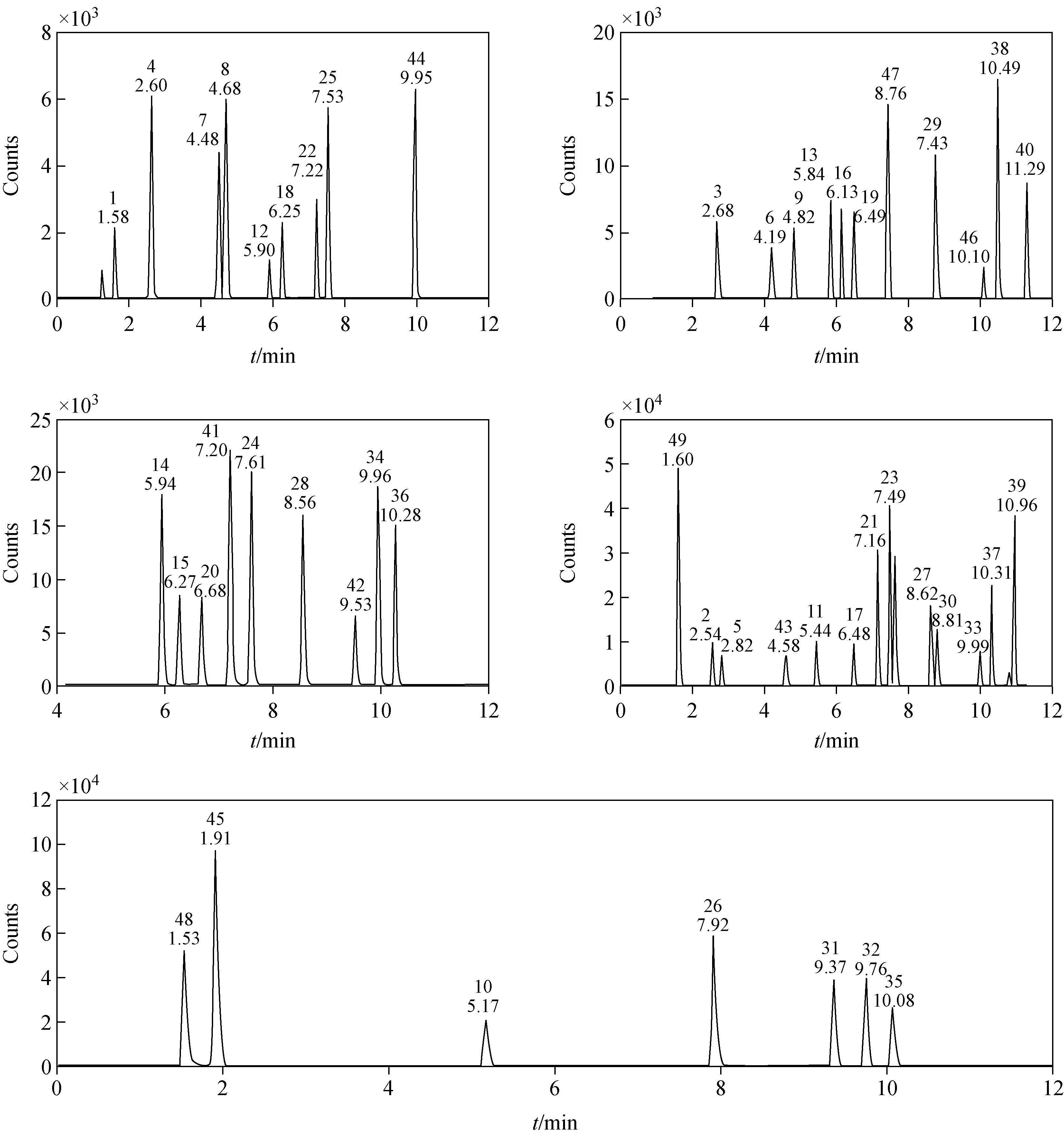
注:峰序号与表1化合物序号一致图2 鸡肉中49种药物的MRM色谱图Fig.2 MRM chromatograms of 49 drugs in chicken tissues
2.6 实际样品分析
利用该方法检测了5批次鸡肉样品,通过比较样品与对照品图谱的保留时间以及监测离子通道的丰度比,判定样品中是否含有糖皮质激素和非甾体抗炎药,通过目标物的质谱响应峰面积与标准对照品比值确定目标物含量。5批样品中有1批次样品检测出氢化可的松药物,含量为1.6 μg/kg,但是氢化可的松在动物性食品中允许使用。该检测结果表明,鸡肉样品中糖皮质激素和非甾体抗炎药的检出率较低。
3 结论
本实验建立了超高效液相色谱-串联质谱法测定鸡肉中的糖皮质激素和非甾体抗炎药,采用Oasis PRIME HLB固相萃取柱,无需活化和平衡步骤,方便快捷,缩短了前处理时间,减少了有机试剂使用量。该方法操作简单、适用性强、准确性好,方法灵敏度能够满足国际上对动物源食品中糖皮质激素和非甾体抗炎药物残留的同时定性、定量检测的要求。
猜你喜欢
中国合理用药探索(2022年4期)2022-11-26
中华养生保健(2020年1期)2020-11-16
科学导报(2019年19期)2019-09-23
中老年保健(2019年3期)2019-03-04
中国现代药物应用(2017年23期)2017-12-16
中西医结合心血管病杂志(电子版)(2015年35期)2015-01-21
中国药业(2014年24期)2014-05-26
中医研究(2013年10期)2013-03-11
