
番茄不同截短U3启动子的克隆及功能分析
2019-03-08 05:49刘晓东阿尔祖古丽塔什
华北农学报 2019年1期
蒲 艳,刘晓东,阿尔祖古丽·塔什,魏 倩,刘 超
(新疆农业大学 农学院,新疆农业大学农业生物技术重点实验室,新疆 乌鲁木齐 830052)
番茄属于茄科,在2012年完成了番茄全基因组测序,其具有较完整的遗传转化体系,这使得番茄成为一个可以很好地运用于科学研究的模式之物。然而,由于缺乏有效的突变体遗传材料,笔者对这些基因或DNA片段的作用或功能却知之甚少。为了从大量的数据中更好地了解基因的功能,迫切需要新的基因编辑技术来满足日益增长的科研需求;同时,通过基因编辑技术培育具有优良性状的农作物可以在一定程度上解决农作物的转基因问题。
近几年,基因编辑技术成为生物学研究的热点,其主要包括锌指核酸酶(Zinc-finger nucleases, ZFNs)[1]、TALEN核酸酶(Transcription activator like effector nucleases, TALENs)[2]、CRISPR/Cas(规律成簇间隔短回文重复序列clustered regularly interspaced short palindromic repeats/CRISPR-associated protein)[3]3种技术体系。而Type Ⅱ的CRISPR/Cas9系统作为一项新型的基因组编辑技术,是继ZFNs及TALEN后在作物基因功能组学研究领域新进的一个基因打靶系统,其载体构建简单,特异性高,据报道已成功地在许多不同的物种中实现了基因的定点编辑[4-12]。CRISPR/Cas9系统中,行使向导功能的是由tracrRNA与crRNA分子间形成的RNA二元复合体改造的单链RNA嵌合体sgRNA,其招募Cas9核酸酶对靶位点进行切割,切割位点于PAM位点上游3~4个碱基处,PAM序列为紧邻靶位点的5′-NGG-3′。而U3启动子是CRISPR/Cas9系统中重要的元件之一,能驱动sgRNA的转录。在植物中,U3和U6启动子由RNA聚合酶Ⅲ转录,其具有非常明确的转录起始位点,并且转录活性较高,U3启动子转录起始位点为A,U6启动子转录起始位点为G,明确的转录起始位点在CRISPR/Cas9系统靶序列的设计及构建更加精确[13-14],同时可以保证不会有多余的序列被转录出来,在sgRNA靶向基因组模板时,也可以提高特异性,从而减少脱靶的现象[15-16],如Shan等[12]利用水稻内源U3启动子驱动sgRNA转录,成功在水稻中实现了PDS和DEP1基因的编辑。Liang等[9]克隆了玉米U3启动子,并成功在玉米原生质体中实现了内源基因ZmIPK的编辑。虽然目前有关U3启动子的CRISPR/Cas9系统在许多物种中已经得到了应用,但相同的U3启动子在同缘关系较远的物种中不一定适用,并且目前未见适用于番茄的内源U3启动子的报道,筛选出在番茄叶片中有功能活性的U3启动子很有必要。USE和TATA框是真核生物中U3启动子发挥转录功能的2个重要元件,本研究通过对候选的3种U3启动子与拟南芥U3启动子序列比对,发现番茄的U3启动子内也含有非常保守的2个元件,但它们是否均具有转录活性还需进一步验证。
本研究从番茄中蔬四号中克隆了3种6个不同长度的U3启动子,再利用农杆菌转化法侵染番茄叶片,通过GUS染色对其转录活性进行分析,筛选出活性较高的U3启动子,旨在为构建番茄CRISPR/Cas9基因编辑载体提供更多高效的内源启动子。
1 材料和方法
1.1 试验材料
试验所用中蔬四号番茄品种、农杆菌GV3101均由新疆农业大学农学院实验室保存。限制性内切酶购于Thermo公司;克隆感受态细胞Trans5α、Blunt Zero平端载体、T4DNA连接酶、1 kb Plus DNA Ladder、植物基因组DNA提取试剂盒、胶回收试剂盒等均购自北京全式金生物技术公司;PhusionDNA聚合酶购于北京NEB公司;引物合成和测序由上海杰李生物科技有限公司完成。
1.2 番茄SlU3-1P、SlU3-3P、SlU3-4P启动子的克隆
将中蔬四号番茄品种播种于含蛭石和营养土的土壤中,取生长21~28 d番茄幼苗叶片,采用植物基因组DNA提取试剂盒提取番茄基因组DNA,用保守的U3 RNA序列在番茄基因组数据库中搜索候选U3 RNA对应的U3启动子序列。本研究采用2轮PCR的方法,以番茄基因组DNA为模板设计引物(表1),第1轮PCR扩增产物为SlU3-1P、SlU3-3P、SlU3-4P启动子全长,用PhusionDNA 聚合酶进行扩增,反应程序为94 ℃ 4 min;98 ℃ 30 s,58 ℃ 30 s,72 ℃ 2 min 30 s,35个循环;72 ℃ 8 min。反应体系为5×Phusion Buffer 4 μL;2.5 mmol/L dNTP 1.6 μL;ddH2O 11 μL;上下游引物各1 μL;Phusion超保真DNA 聚合酶0.4 μL;DNA模板1 μL;共20 μL体系。
第2轮扩增的受体质粒为GhU6-5P2::GUS,供体质粒为SlU3-1P、SlU3-3P、SlU3-4P启动子进行Transfer PCR[17],根据第1轮测序正确的SlU3-1P、SlU3-3P、SlU3-4P启动子序列和GhU6-5P2::GUS载体序列分别设计了2对引物(表1)。使用超保真DNA 聚合酶进行扩增,反应程序为94 ℃ 1 min;94 ℃ 30 s,60 ℃ 1 min,72 ℃ 90 s,13个循环;94 ℃ 30 s,67 ℃ 1 min,72 ℃ 4 min,20个循环;72 ℃ 8 min。反应体系为5×Phusion Buffer 10 μL;2.5 mmol/L dNTP 2.5 μL;上下游引物各1 μL;供体质粒和受体质粒模板各1 μL;ddH2O 32.7 μL;Phusion超保真DNA 聚合酶0.8 μL;共50 μL体系。通过琼脂糖凝胶电泳对PCR产物进行分析,对分析正确的PCR产物用DpnⅠ在37 ℃进行酶切处理3 h,取酶切产物10 μL转化Trans5α感受态细胞,用BglⅡ和NotⅠ双酶切鉴定SlU3-1P重组质粒,XhoⅠ和NotⅠ双酶切鉴定SlU3-3P重组质粒,NsiⅠ和NotⅠ双酶切鉴定SlU3-4P重组质粒,酶切鉴定后选取正确的质粒进行测序,测序正确的质粒命名为T-SlU3-Ps。

表1 引物序列Tab.1 Primer sequences
注:下划线部分为受体质粒碱基序列。
Note:Underlined for the acceptor plasmid partial base sequence.
1.3 SlU3启动子序列分析
运用 DNAMAN 软件对候选的番茄U3启动子与拟南芥U3启动子进行序列比对,对候选的U3启动子内功能元件进行分析。
1.4 不同截短SlU3Ps∷GUS-sgRNA-P1300融合表达载体的构建
用KpnⅠ和HindⅢ双酶切植物表达载体 pCAM-BIA 1300及SlU3-1P、SlU3-3P、SlU3-4P后分别回收目标片段并用T4连接酶连接、转化后对重组质粒进行酶切鉴定,鉴定正确的质粒命名为SlU3∷GUS-sgRNA-P1300,转入农杆菌感受态细胞GV3101后,于28 ℃倒置培养2 d,挑取阳性克隆于LB培养基中(含 Rif 25μg/mL和Kan 50μg/mL)培养至对数生长期。不同SlU3Ps∷GUS-sgRNA-P1300 融合表达载体示意图如图1所示。
1.5 农杆菌介导法转染番茄叶片
将构建成功的不同SlU3-P∷GUS-P1300以及阳性对照P1301和阴性对照pCAMBIA1300农杆菌,按照1∶100比例接种于LB培养基(含50 μg/mL Kan和25 μg/mL Rif)中180 r/min,28 ℃至菌液OD600=1时,分别吸取菌液到5 mL LB培养基(含50 μg/mL Kan和25 μg/mL Rif)中,每个菌液的OD值相同后,再次28 ℃, 180 r/min至所有菌液的OD600=1.5,12 000 r/min离心5 min收集菌体于重悬Buffer(50 mmol/L MgCl2、200 mmol/L MES、20 mmol/L乙酰丁香酮)至1 mL,室温放置3 h后分别注射到生长21 d的番茄子叶中,最后进行过夜暗培养。

图1 不同截短SlU3-Ps启动子驱动报告基因GUS的融合表达载体构建Fig.1 Construction of GUS reporter gene fusion expression vector driven by different truncated SlU3-Ps promoter
1.6 不同截短SlU3-Ps∷GUS在番茄叶片中的瞬时表达分析
剪下暗培养过夜后的番茄叶片浸泡在150 μLGUS染液中(0.5 mol/L EDTA, pH值8.0;0.5 mol/L磷酸缓冲液, pH值7.0;20 mmol/L X-Gluc;10% Triton X-100),-0.05 MPa下抽真空30 min。抽完真空后置于37 ℃,180 r/min振荡染色6~7 h后100 r/min离心1 min吸取上清GUS染液。为彻底去除残留的GUS染液,用蒸馏水漂洗染色后的叶片4~5次,100 r/min 离心1 min。染色完成后加入300 μL无水乙醇进行脱色处理,每3~4 h更换一次脱色液,直至叶片呈透明状。
2 结果与分析
2.1 第1轮SlU3-1P、SlU3-3P、SlU3-4P启动子扩增产物鉴定
从中蔬四号番茄中成功扩增出SlU3-1P、SlU3-3P、SlU3-4P启动子片段,长度分别为1 156,1 114,1 147 bp(图2)。用BglⅡ和NotⅠ双酶切鉴定SlU3-1P重组质粒,XhoⅠ和NotⅠ 双酶切鉴定SlU3-3P重组质粒,NsiⅠ和NotⅠ双酶切鉴定SlU3-4P重组质粒,通过琼脂糖凝胶电泳检测结果正确,最终测序比对后为预期的SlU3-1P、SlU3-3P、SlU3-4P启动子。
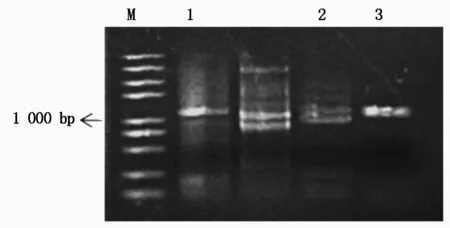
M.2 kb PlusⅡ标准分子量;1.SlU3-1P,1 156 bp;2.SlU3-3P,1 114 bp;3.SlU3-4P,1 147 bp。M.2 kb PlusⅡDNA Marker; 1.SlU3-1P,1 156 bp; 2.SlU3-3P,1 114 bp; 3.SlU3-4P,1 147 bp.
2.2 Transfer PCR扩增产物鉴定
以测序正确的第1轮SlU3-1P、SlU3-3P、SlU3-4P为供体质粒,GhU6-5P2∷GUS受体质粒进行Transfer PCR,其目的是将受体质粒GhU6-5P2∷GUS启动子片段置换为SlU3∷GUS-sgRNA片段。图3为成功克隆的不同截短的T-SlU3-1P4f、T-SlU3-1P5f、T-SlU3-3P4f、T-SlU3-3P5f、T-SlU3-4P4f、T-SlU3-4P5f启动子电泳图,其片段大小依次是489,318,450,248,457,248 bp。用MfeⅠ和Hind Ⅲ分别双酶切鉴定各截短启动子质粒,电泳后发现,酶切结果与预测片段大小一致,测序比对后为预期截短的T-SlU3Ps启动子(图3)。
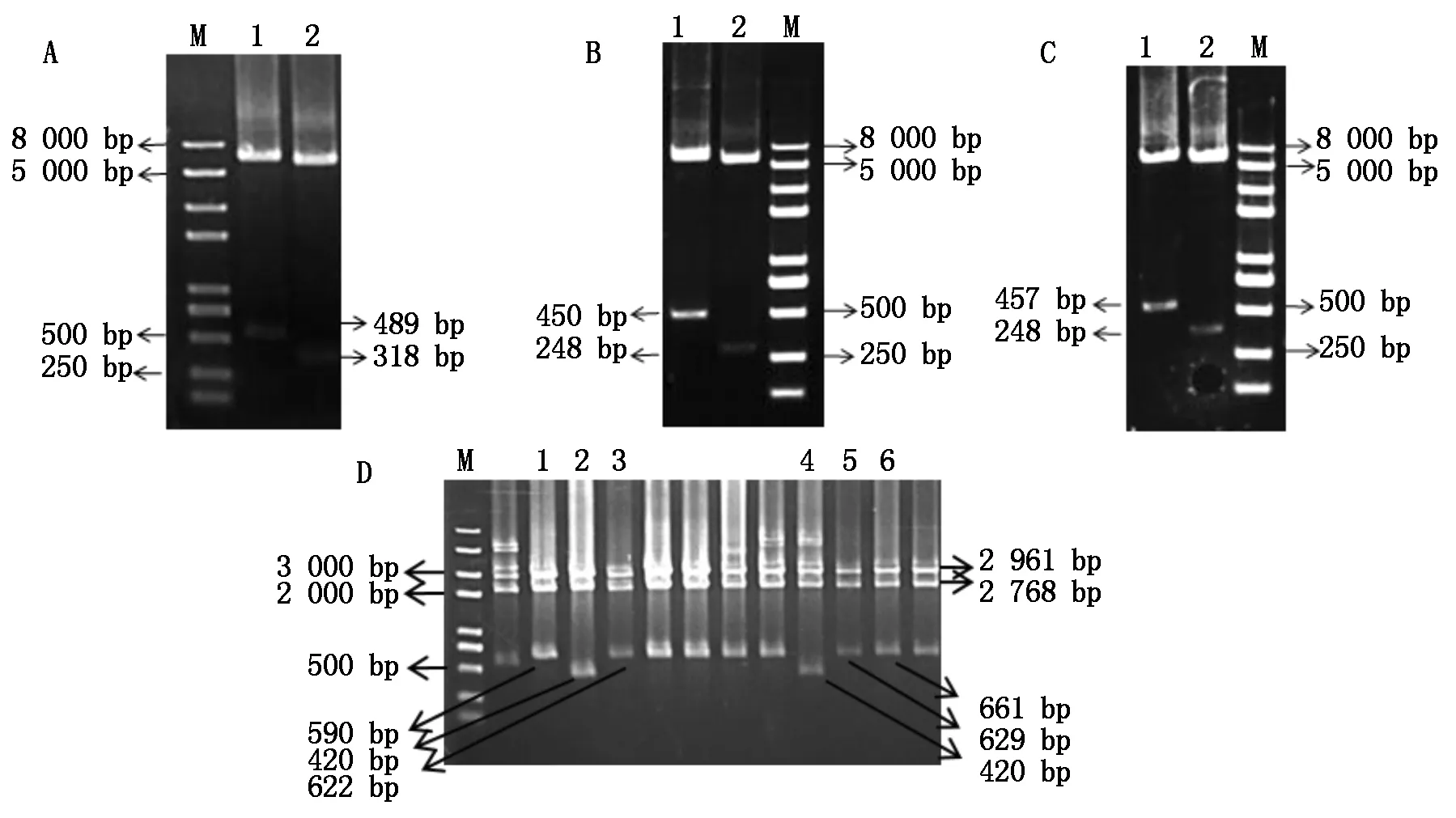
A、B、C.Transfer PCR扩增;M. 2 kb PlusⅡ DNA标准分子量;A: 1.T-SlU3-1P4f;2.T-SlU3-1P5f; B: 1.T-SlU3-3P4f;2.T-SlU3-3P5f;C: 1.T-SlU3-4P4f;2.T-SlU3-4P5f;图中小片段为第1阶段扩增的SlU3启动子,大片段为第2阶段扩增产物。D.酶切鉴定: 1.T-SlU3-1P5;2.T-SlU3-3P5;3.T-SlU3-3P4;4.T-SlU3-4P5;5.T-SlU3-4P4;6.T-SlU3-1P4.
A,B,C.Transfer PCR amplificaton;M. 2 kb PlusⅡDNA Marker; A: 1.T-SlU3-1P4f;2.T-SlU3-1P5f;B:1.T-SlU3-3P4f;2.T-SlU3-3P5f; C:1.T-SlU3-4P4f;2.T-SlU3-4P5f;The small fragment in the figure is the first-stage amplifiedSlU3 promoter, and the large fragment is the second-stage amplification product.D. Identification: 1.T-SlU3-1P5; 2.T-SlU3-3P5; 3.T-SlU3-3P4;4.T-SlU3-4P5; 5.T-SlU3-4P4; 6.T-SlU3-1P4.
图3三种番茄U3启动子的TransferPCR扩增及酶切鉴定
Fig.3TransferPCRamplificationandidentificationofthreekindsoftomatoU3promoters
2.3 SlU3启动子序列分析
对本研究已克隆的3种番茄U3启动子和拟南芥U3启动子进行序列比对,发现番茄U3启动子区域包含RNA聚合酶Ⅲ转录所需要的元件,位于-30 bp的TATA框和位于-60 bp的USE元件5′-RTCCCA CATCG-3′,图中方框为USE和TATA框,黑色箭头处为U3 RNA的起始位点(图4)。

图4 SlU3启动子序列分析Fig.4 Sequence analysis of SlU3 promoters
2.4 不同截短SlU3Ps∷GUS-sgRNA-P1300融合表达载体构建
将正确的番茄不同截短SlU3Ps∷GUS-sgRNA的重组质粒片段连入植物表达载体pCAMBIA1300,用KpnⅠ和HindⅢ分别对不同SlU3Ps∷GUS-sgRNA-P1300重组质粒进行酶切鉴定,酶切的结果和预测片段大小一致(图5)。
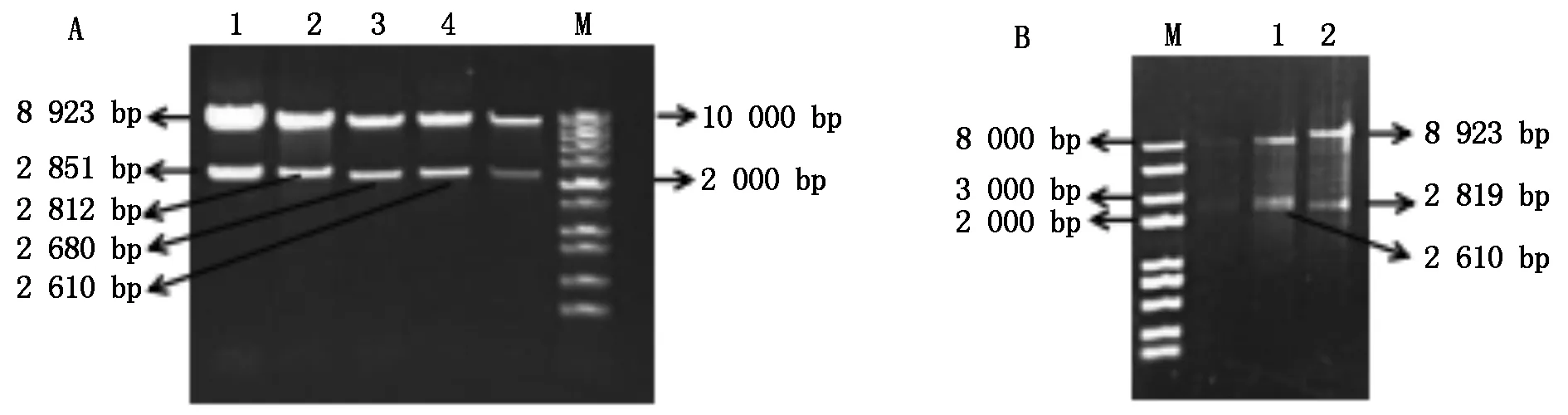
A: M.1 kb DNA标准分子量;1.SlU3-1P4∷GUS-sgRNA-P1300;2.SlU3-3P4∷GUS-sgRNA-P1300;3.SlU3-1P5∷GUS-sgRNA-P1300;4.SlU3-3P5∷GUS-sgRNA-P1300;B:M.2 kb plusⅡ标准分子量;1.SlU3-4P4∷GUS-sgRNA-P1300;2.SlU3-4P5∷GUS-sgRNA-P1300。A:M.1 kb DNA Marker;1.SlU3-1P4∷GUS-sgRNA-P1300; 2.SlU3-3P4∷GUS-sgRNA-P1300; 3.SlU3-1P5∷GUS-sgRNA-P1300;4.SlU3-3P5∷GUS-sgRNA-P1300;B:M.2 kb plusⅡ DNA Marker; 1.SlU3-4P4∷GUS-sgRNA-P1300; 2.SlU3-4P5∷GUS-sgRNA-P1300.
2.5 不同截短SlU3启动子功能分析
将构建好的不同截短SlU3Ps∷GUS-sgRNA-P1300,阴性对照pCAMBIA1300及阳性对照pCAMBIA1301通过农杆菌介导法注射番茄叶片。通过GUS组织化学染色分析结果显示,侵染成功后的番茄叶片均被染成蓝色,其表明已克隆的6种SlU3启动子均能驱动报告基因GUS的表达,但其表达水平存在一定差异(图6)。

图6 番茄不同截短SlU3启动子驱动GUS在番茄叶片中的瞬时表达Fig.6 Transient expression of GUS in tomato leaves driven by different truncated SlU3 promoters in tomato
3 讨论
Ⅱ型CRISPR/Cas9基因组编辑系统作为一项全新的基因组定点修饰技术,近几年已被成功地应用于不同植物中,能实现在基因组特定位点的修饰。在基因组编辑的过程中,sgRNA能够靶向结合到目的基因位点,与CRISPR/Cas9系统的特异性相关[18]。其中U3启动子是重要组成成分之一,被广泛用于驱动sgRNA的转录。U3启动子具有非常明确的转录起始位点(A)且序列较短,其在生物体内驱动的U3 snRNA序列非常保守。依据这些信息U3启动子可以被精确的克隆。在真核生物体内存在多种U3启动子,每个启动子是否均具有转录活性并不清楚。虽然有关U3启动子的CRISPR/Cas9系统已在许多物种中得到了应用,但同样的U3启动子在同缘关系较远的物种中不一定适用[19],并且目前番茄内源U3启动子的研究还未见报道,因此,筛选出在番茄中具有功能活性的U3启动子很有必要。另外,虽然刘晓东课题组已克隆了番茄的U6启动子,并且利用该启动子将番茄内源基因成功地实现了编辑,建立了番茄的CRISPR/Cas9基因组编辑技术体系。但由于研究的需要,如研究一个基因家族的功能、多基因控制的复杂数量性状的研究、单个基因多位点编辑以及长基因片段的删除,都需要克隆出更多U3或U6启动子,用于构建多位点基因编辑载体。如拟南芥PYL和水稻FTL基因家族成员的多基因敲除,都使用了植物内源多种U3和U6启动子[16]。
番茄中至少含有4个U3启动子,本课题组克隆的3个U3启动子,将它们和拟南芥U3启动子进行序列比对分析,发现番茄U3启动子区域包含RNA聚合酶Ⅲ转录所需要的USE元件和TATA框[20],且这2个元件的序列非常保守、它们之间的距离也比较固定,则推测番茄U3启动子也同样具有转录活性。为了方便构建CRISPR/Cas9基因组编辑载体,所用的U3启动子对其转录活性和长度上都有很高的要求,因此,有必要克隆长度适宜且活性较高的番茄U3启动子。本研究通过番茄不同U3启动子驱动GUS报告基因,并通过农杆菌介导法瞬时转化番茄叶片来验证其功能活性。通过GUS组织化学染色分析结果显示,侵染成功后的番茄叶片均被染成蓝色,表明克隆的6种U3启动子均能驱动报告基因GUS的表达,都具有转录活性,当这3种启动子截短到300 bp左右,甚至是250 bp以内的长度时,仍然能驱动报告基因GUS的表达。这显示出番茄的U3启动子也相对较短,与在其他作物中克隆的U3启动子特点相同[21]。这些启动子进一步截短后是否还具有转录活性,为了构建多位点基因编辑载体,也需要在后续的工作中对进一步截短的启动子的功能进行研究。本研究克隆的启动子可为后期在番茄中建立高效的CRISPR/Cas9基因组编辑载体提供更多适用于驱动sgRNA的启动子。
猜你喜欢
环球时报(2022-09-20)2022-09-20
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
军事文摘(2022年16期)2022-08-24
成都医学院学报(2022年4期)2022-08-19
今日农业(2021年11期)2021-08-13
江西农业学报(2021年4期)2021-04-20
今日农业(2020年24期)2020-12-15
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
三农资讯半月报(2020年11期)2020-06-21
