
HPLC-MS/MS法测定人血浆中伏立康唑浓度的不确定度评价
2019-01-30 06:49于思源冯志平
中国抗生素杂志 2019年1期
于思源 冯志平
(江门市中心医院药学部,中山大学附属江门医院,江门 529000)
不确定度的定义为表征合理地赋予被测量之值的分散性,广义而言,意为对测量结果正确性的可疑程度。不确定度的评定方法通常有A和B两种,A类评定是由一系列重复观测值计算得到,而B类评定是根据有关信息(假定的概率密度函数)来评定[1]。伏立康唑作为新一代三唑类广谱抗真菌药广泛用于治疗血液病、移植或免疫缺陷患者的侵袭性真菌感染[3-4]。由于伏立康唑主要通过细胞色素P450同工酶2C19酶(CYP2C19)代谢,该酶活性存在显著的个体和种族差异,表现为遗传多态性[5-7];不同人群间病理生理差异等也可能使伏立康唑体内药动学特征产生较大差异,从而导致个体间血药浓度偏差较大,浓度过高易发生不良反应(ADR),而浓度过低则导致临床治疗失败。研究表明伏立康唑的血药浓度与临床疗效、ADR的关系密切[8]。因此浓度检测的准确性是准确调整用药的前提。目前为止,伏立康唑液质联用测定方法的不确定度尚未见文献报道,为了评估测定方法的准确性,改进实验步骤,提高检测质量,本文根据相关的规范和指南[1-2]并参考相关文献[9-12],对HPLC-MS/MS测定人血浆中伏立康唑浓度的不确定度进行评价。
1 仪器与试药
1.1 仪器
Agilent 1290高效液相色谱仪、6460C三重四级杆串联质谱仪,美国Aglient科技公司;高速冷冻离心机,德国Sigma公司;XSE205DU型电子分析天平,瑞士Mettler公司;GenPure,美国Thermo Fisher Scientific公司。
1.2 试药
伏立康唑对照品(批号:81607441,质量分数:99.50%,珠海丽珠制药厂);氯雷他定对照品(内标,批号:DC-0201-1503002,质量分数:99.80%,浙江东亚药业股份有限公司);乙腈和甲醇均为色谱纯,Thermo Fisher Scientific公司;甲酸为色谱纯,Dikma Technologies公司;甲酸铵为色谱纯,Sigma-Aldrich公司。方法学研究所用健康人空白血浆,由江门市中心医院输血科提供。
2 方法与结果
2.1 色谱-质谱条件
色谱柱为Agilent ZORBAX Eclipse XDB-C18柱(50mm×4.6mm, 1.8μm);流动相比例为乙腈:含0.1%甲酸的10mmol/L甲酸铵水溶液(85:15),等度洗脱2.5min,流速为0.4mL/min,柱温为40℃,进样量为1μL。
采用电喷雾离子源(ESI),正离子MRM模式,质谱参数如下:毛细管电压4000V,干燥气流速11L/min,雾化气压力15psi,干燥气温度300℃,伏立康唑:碎裂电压120V,碰撞能量15V,离子对:m/z350.0→280.9;氯雷他定:碎裂电压140V,碰撞能量20V,离子对:m/z383.0→337.0。
2.2 溶液的制备
精密称取伏立康唑对照品10.17mg,置于5mL容量瓶中,50%甲醇溶解并定容,摇匀,得浓度为2.02mg/mL的伏立康唑储备液,置于4℃冰箱中保存,备用。临用前取伏立康唑储备液适量,用50%甲醇逐级稀释成质量浓度为0.2、0.4、2、5、20、50、100和200μg/mL的系列标准溶液,置于5mL容量瓶中;另取伏立康唑储备液适量,用50%甲醇逐级稀释成质量浓度为0.5、30和160μg/mL的质控溶液置于5mL容量瓶中。
精密称取内标对照品11.50mg,置于5mL容量瓶中,50%甲醇溶解并定容,摇匀,得浓度为2.295mg/mL的内标储备液,置于4℃冰箱中保存,备用。临用前将内标储备液用50%甲醇稀释成浓度为3μg/mL的内标溶液,置于5mL容量瓶中。
2.3 标准曲线样品和质控样品的配置、处理
取95μL空白人血浆,加入伏立康唑系列标准溶液和质控溶液各5μL,配制成质量浓度为0.01、0.02、0.1、0.25、1.0、2.5、5和10μg/mL的标准曲线血浆样品及0.025、1.5和8.0μg/mL的质控样品。
精密吸取含药血浆样品100μL,加入3μg/mL内标溶液5μL,涡旋1min;再加入纯乙腈600μL沉淀蛋白,涡旋振荡3min,15000r/min离心5min,取其上层清液100μL转移至进样瓶内,进样1μL分析。
2.4 数学模型的建立
以VOR浓度为x轴,以VOR与内标信号的面积比为y轴,用最小二乘法进行线性回归,线性回归方程:y=ax+b,其中:x为VOR浓度,y为VOR与内标的峰面积比,a为回归方程的斜率,b为截距。
2.5 测量不确定度来源分析
分析整个实验流程,测量不确定度的来源主要有:温度、重复性、对照品称量、工作液配制、样品制备、基质效应、样品提取、标准曲线拟合、仪器允差、对照品纯度和样品不均匀性等因素(图1)。
2.6 温度对测定的影响
实验室温度控制在(20±2)℃,标准曲线和待测样品均在相同温度下制备和测定,因此温度引入的不确定度可忽略。
2.7 测量不确定度的评定
2.7.1 重复性(用A类评定程序)
重复性引入的不确定度用质控样品重复测定值评估,低、中、高3个浓度(0.025、1.5和8.0μg/mL)的质控样品各3组(m=3),每组平行测量5次(n=5),结果见表1。用贝塞尔公式计算合并样品偏差:
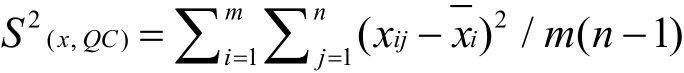
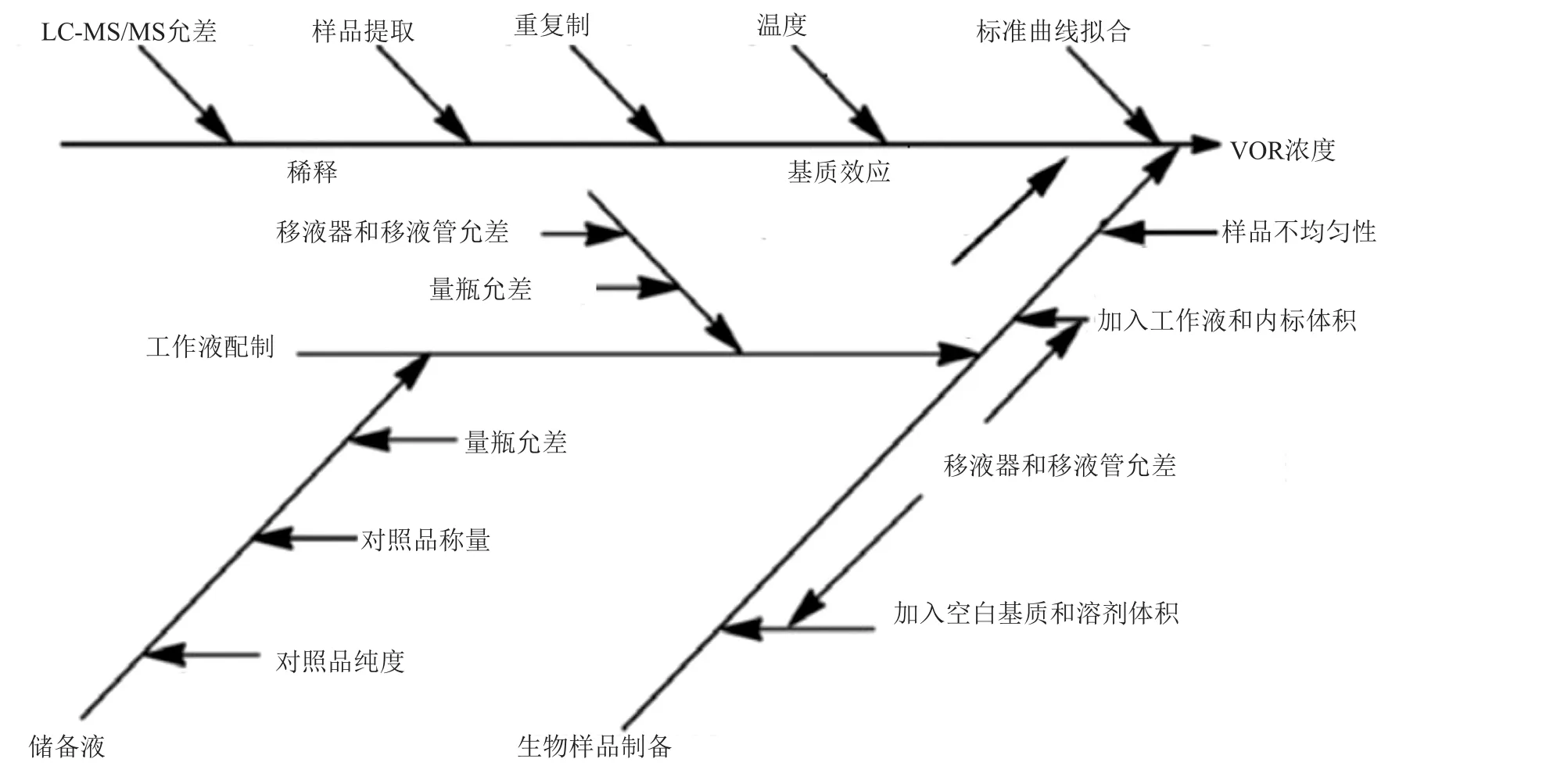
图1 不确定度来源的因果分析图Fig.1 Causal analysis chart of uncertainty sources

表1 样品重复测定数据Tab. 1 Repeatability of sample determination
其中:i为组数,j为每组平行测定次数(i=3,j=5)。每组5个测定值的平均值的标准偏差为:
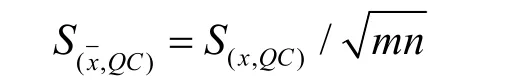
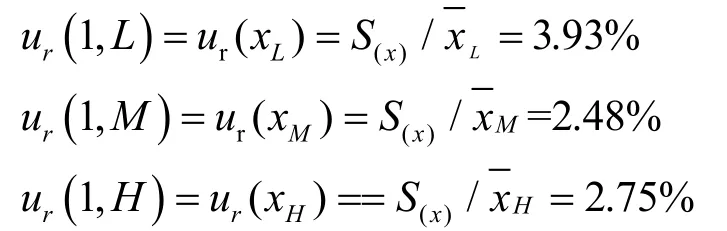
2.7.2 对照品称量引入的不确定度(用B类评定程序)
对照品称量引起的不确定度可表示为:
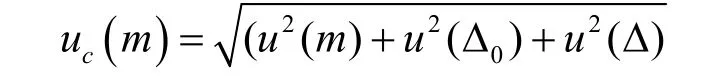
其中,天平的重复性误差u(m)在重复性实验中已经得到评定,不再重复计算。由自动调零引起的误差u(△0)和天平的非线性误差u(△)按均匀分布,包含因子k=3,随机变量半宽a=0.5e,依据计量检定证书,天平检定分度值e=0.01mg,则标准测量不确定度为:
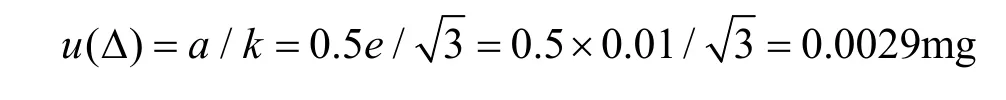
减重对照品称量时自动调零作为一次扣皮,则a0=a,u(△0)=a0/k=u(△)=0.0029。天平的标准测量不确定度为:
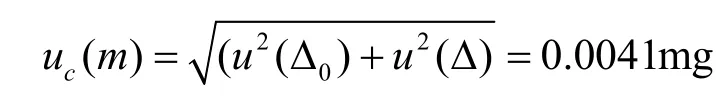
VOR对照品称量的质量为10.17mg,则VOR对照品称量相对标准测量不确定度为:
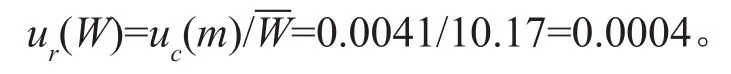
内标(IS)对照品称量的质量为11.50mg,则内标对照品称量相对标准测量不确定度为:
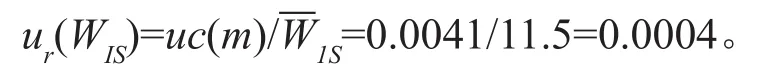
称量引起的相对标准测量不确定度为:
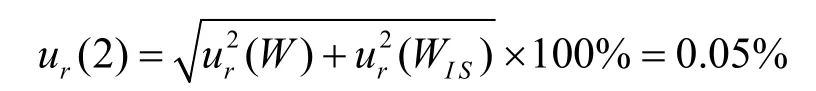
2.7.3 样品配制引入的测量不确定度(用B类评定程序)
(1)容量瓶引入的测量不确定度:用于储备液配制的A 级容量瓶(F)规格为5mL,其最大允许误差为±0.020mL。由于温度对定容的影响非常小,忽略不计。按均匀分布,其相对标准不确定度为:ur=a/(kx),则:
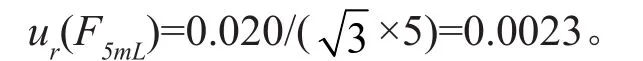
(2)移液器引入的测量不确定度:本次所用移液器为Eppendorf可调量程移液器,其中所用的Eppendorf移液器量程为:0.5~10μL(P1),10~100μL(P2)、100~1000μL(P3)和0.5~5μL(P4)。参照Eppendorf说明书,P1在5μL时最大允差为±1.5%;P2在100μL时最大允差为±0.8%;P3在500和1000μL时最大允差分别为±1%、±0.6%;P4在2~2.5mL时最大允差为±1.2%。按均匀分布,包含因子,则移液器的相对标准测量不确定度:ur(P1-5)=0.0061,ur(P2-100)=0.0033,ur(P3-500)=0.0033,ur(P3-1000)=0.0024,ur(P4-2.5)=0.0049。
(3)标准溶液和质控溶液配制引入的测量不确定度:储备液配制时的相对标准不确定度已经在容量瓶定容中引入,因此可以忽略;在使用储备液配制标准溶液(STD)时,共使用5mL容量瓶8次;100~1000μL(P3)移液器2次,其中使用500μL 1次,1000μL 1次;0.5~5mL(P4)移液器3次,其中使用2~2.5mL 6次;在使用储备液配制质控溶液(QC)时共使用5mL容量瓶4次,低质控2次,中质控1次,高质控1次;100~1000μL(P3)移液器4次,其中高质控使用100~500μL 1次,中质控使用500~1000μL 1次,低质控使用500~1000μL 2次。查阅相关文献[13],标称容量为介于给定的两个容量点之间的值,那么它的最大允差是以任一两个容量之间较大容量为标准的,因此100~500μL的最大允差按500μL计算,500~1000μL的最大允差按1000μL计算,2mL的最大允差按2.5mL计算。内标配制不影响测定,则对照品溶液配制引入的相对标准测量不确定度为:

(4)含药标准样品和质控样品配制引入的不确定度:配制含药标准样品(S)和质控样品(QC)时,使用10~100μL移液器吸取95μL空白血浆,后使用0.5~10μL移液器吸取系列标准溶液或质控溶液、内标溶液各5μL,加入到空白血浆中,后使用100~1000μL移液器加入600μL纯乙腈进行样品提取。因此,所用移液器的型号和次数均为:0.5~10μL(P1)2次,10~10μL(P2)1次,100~100μL(P3)1次,同上“2.7.3”项“(3)”中最大允差计算原则,则配制含药标准样品和质控样品的相对标准测量不确定度为:
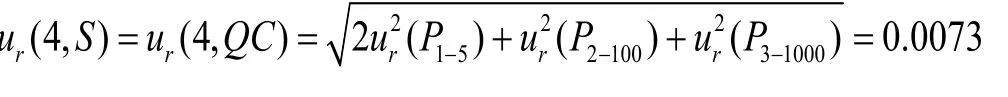
则样品配制过程中引入的相对不确定度为:
该段落第二句是论据句,它只是呈现了一个事实:歌手庞麦郎以一首《我的滑板鞋》惊醒了很多当代人。而通过分析,我们恍然大悟:清醒地与时代保持一段距离其实也是真实地表现时代的声音,这样的声音恰恰赢得时代的共鸣。这样的论据虽然只有一个,但很有说服力。很多考生错误地认为,论据越多越好,其实不然,精当的论据常起到一以当十的效果。
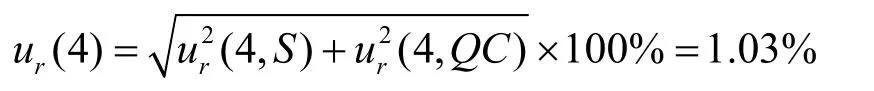
2.7.4 基质效应和蛋白沉淀过程引入的测量不确定度(用A类评定程序)
(1)基质效应的影响:质谱法中,基质效应(ME)可影响最终的测定结果,其不确定度必须考虑。评估方法如下:分别用处理后基质及空白溶剂配制低、高2个浓度(0.025和8.0μg/mL)质控样品,平行测定5次,计算方法:基质效应=处理后基质配制样品的峰面积与内标峰面积比值(A)/空白溶剂配制样品的峰面积与内标峰面积比值(B),经内标校正后的低浓度和高浓度质控血浆样品的基质效应因子分别为(90.68±2.94)%及(93.31±3.18)%,则基质效应ur(5)引入的相对标准测量不确定度为:
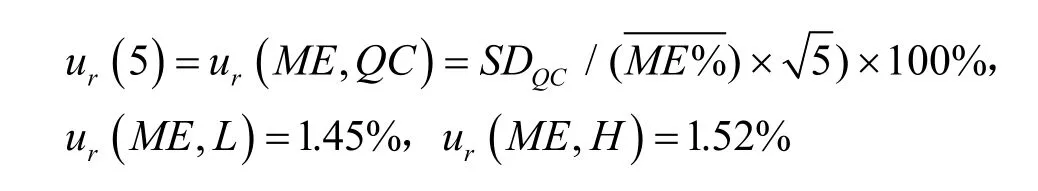
(2)样品提取过程的影响:
样品提取过程(RE)同样会影响测定结果,其引入的不确定度由样品提取评估。评估方法如下:以未处理基质和处理后基质制备低、中、高3个浓度(0.025、1.5和8.0μg/mL)的质控样品,每个浓度平行配制3份,计算方法:样品提取(%)=100%×血浆配制样品蛋白沉淀后的峰面积与内标峰面积比值(C)/处理后基质配制样品的峰面积与内标峰面积比值(D),经内标校正后的低、中、高浓度质控血浆样品的样品提取分别为(104.73±10.62)%、(102.47±7.97)%、(94.94±3.67)%,当样本量较小时,样品提取ur(6)引入的相对标准测量不确定度使用极差法进行计算[1],
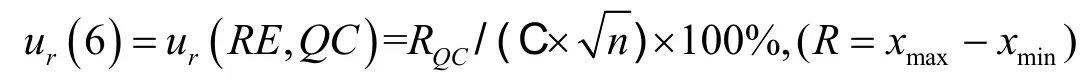
即:
结果分别为:ur(RE,L)=6.07%,ur(RE,M)=4.76%,ur(RE,H)=2.31%。
2.7.5 仪器量化引入的测量不确定度(用B类评定程序)
质谱仪为Agilent 6460C三重串联四极杆质谱仪,液相为Agilent 1290 infinity,根据设备校准报告,进样峰面积重复性为4.9%,按均匀分布,包含因子,仪器量化相对标准测量不确定度为:ur(7)=2.83%。
标准曲线包括8个浓度,用VOR峰面积与内标峰面积的比值对VOR浓度进行标准曲线拟合,以1/x2作为权重(w)。标准曲线中VOR与内标的峰面积比及3条标准曲线的斜率和截距见表2,用拟合的标准曲线反算出对照品血浆的VOR浓度见表3。

表2 VOR与内标峰面积比及回归曲线参数Tab. 2 Area ratio of VOR with the internal standard and parameters of the calibration curves
标准曲线有8个浓度点,n=8,对照品血浆每个浓度测定的次数为3次,m=3,N为测定标准血浆溶液的总次数,N=m×n=24;am为标准曲线的斜率;b为标准曲线截距;xi为第i个标准血浆溶液的浓度,x为8个标准血浆浓度的理论平均值,x=2.3783μg/mL;i为每组对照溶液的序数(i=1, 2, …,m),j为测定血浆标准溶液的序数(j=1, 2 ,3, …,n)。
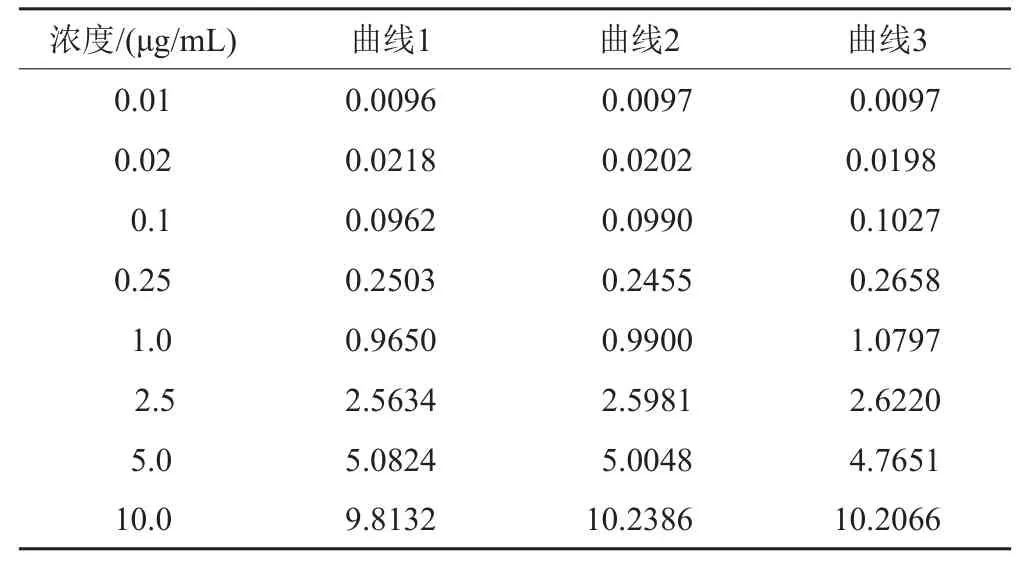
表3 用拟合曲线计算出每个对照品血浆VOR的浓度Tab. 3 Back-calculated concentrations of VOR in standard plasma samples
自相关方差为:
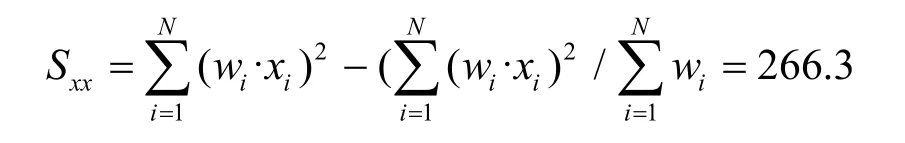
残余标准差为:
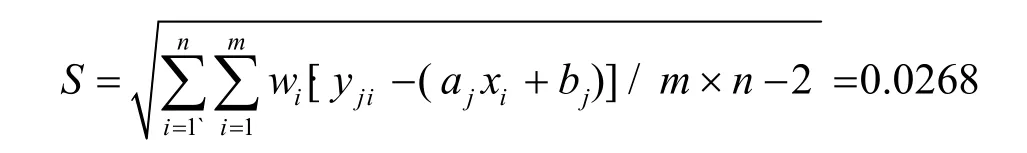
测定低、中、高浓度质控样品各15次,P=15,计算得平均浓度:则标准曲线拟合的标准测量不确定度为:
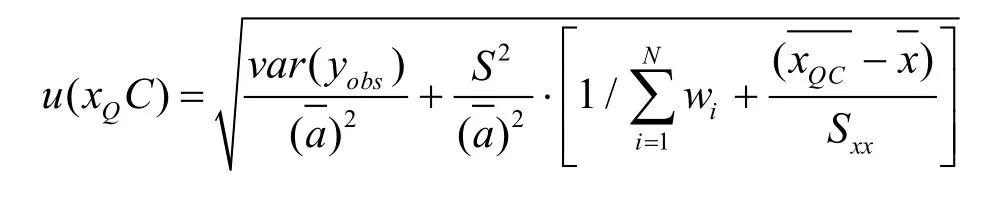
其中,Sxx表示自相关方差,wi为权重。var(yobs)表示样品观测值方差,计算公式:
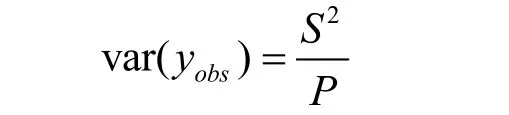
其中,S为残余标准差。u(xL)=0.0082,u(xM)=0.0075,u(xH)=0.0117。则标准曲线拟合引入的相对标准测量不确定度为:
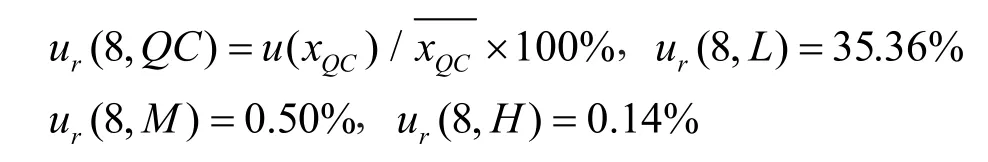
2.7.7 对照品纯度和样品不均匀性引入的不确定度
伏立康唑对照品和内标氯雷他定的质量分数均不是100%,限度标识可按1%计(假定符合均匀分布),则相对不确定度为ur(9)=1%/=0.58%;由于样品均为液态,使用前混合均匀,由样品不均匀性引入的不确定度忽略不计。
2.8 标准测量不确定度的合成及扩展
2.8.1 标准测量不确定度的合成
依据不确定度传播规律对各相对标准测量不确定度进行合成:
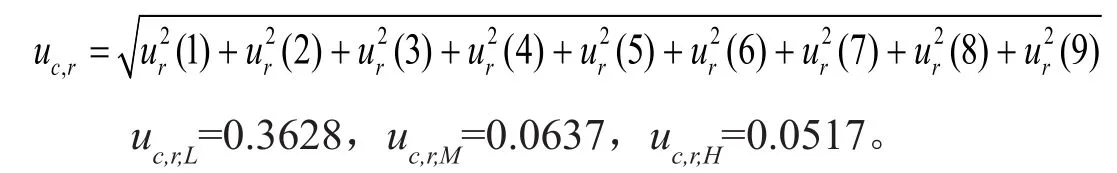
则VOR质控样品的合成标准测量不确定度分别为:
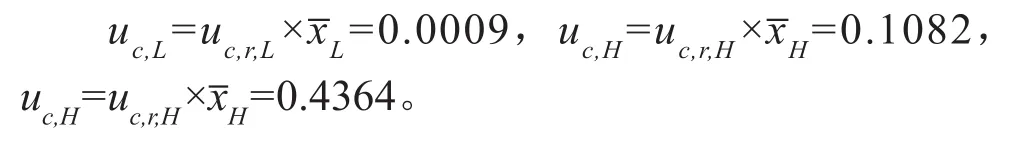
2.8.2 标准测量不确定度的扩展
采用简易评定法,对应的置信概率P=95.45%(k=2),则扩展不确定度分别为:
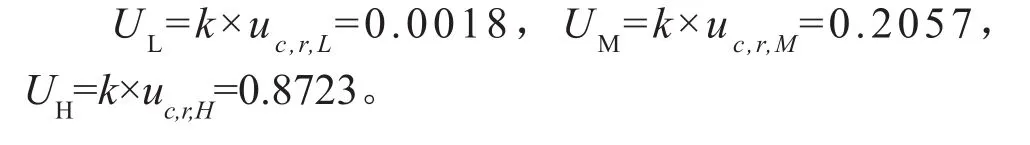
2.9 测定结果的表示
当置信概率P=95.45%(k=2),血浆中VOR低、中、高浓度质控的测定结果可以分别表示为(0.025±0.0018)、(1.5±0.2057)和(8±0.8723)μg/mL。
3 讨论
本文通过HPLC-MS/MS法测定血浆中伏立康唑浓度的实验流程,寻找不确定度的来源并计算,对各不确定度分量进行合成并扩展(图2)。本试验未考虑温度引入的不确定度,原因是实验室温度控制在(20±2)℃,且标准曲线和待测样品几乎在同一时间配制,温度变化很小。样品在制备时均混合均匀,因此样品不均匀性引入的不确定度可忽略不计。
首先,从计算结果可知,对照品称量引入的不确定度很小,与文献报道类似[9-10]几乎可以忽略不计,这是因为对照品称量所用分析天平为十万分之一级,因此配备高精度的天平可显著减小对照品称量引入的误差;高,中、低质控浓度样品提取引入的不确定度差别很大,尤其是低质控的不确定度相对较大,笔者认为主要是由于向处理后的基质中添加内标和质控工作液时未充分混匀或混匀效果不佳,导致测定结果的标准差相对较大。因此,笔者建议在向处理后的基质中添加内标或质控工作液后,轻微涡旋5~10s,溶液扩散应更为均匀,人为操作误差可能更小;标准曲线拟合对低浓度质控溶液的影响最大,可能与标准曲线范围相对较大,测定点的个数和重复测定次数相对较少密切相关;液质联用仪的进样重复性引入的不确定度,笔者建议在正式进样前使用当天一批中最低浓度样品和中等浓度样本,至少各进3针,可能会有效防止质谱信号漂移,减少测量结果的不确定度。
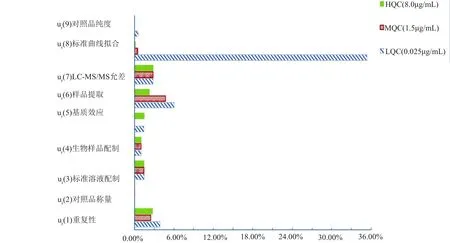
图2 不确定度分量的统计条形图Fig.2 Statistical bar graph of uncertainty components
综上所述,本文评定了LC-MS/MS法测定人血浆中VOR浓度的不确定度,在低浓度时主要由标准曲线拟合,样品提取和重复性引入,在中浓度时主要由样品提取和仪器允差引起,在高浓度时主要由仪器允差,重复性和样品提取引入,另外,依照《中国药典2015年版生物样品定量分析方法验证指导原则》,本次研究在考察基质效应时未将中质控溶液纳入,因此,基质效应对中质控溶液的影响并未评价。
猜你喜欢
中国医院用药评价与分析(2022年3期)2022-04-07
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
中国应急管理科学(2021年4期)2021-04-13
药学服务与研究(2020年4期)2020-09-03
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
中国抗生素杂志(2019年7期)2019-07-25
健康大视野(2019年10期)2019-05-10
中国科技纵横(2019年23期)2019-02-14
