
一种不易诱发耐药性的新型抗生素teixobactin的研究进展
2019-01-30 06:49张焕云李瑞娟
中国抗生素杂志 2019年1期
张焕云 李瑞娟
(山东大学微生物技术国家重点实验室,山东大学-亥姆霍兹生物技术研究所,青岛 266237)
近些年,随着抗生素的广泛使用甚至滥用,细菌耐药性逐渐增加,耐药性细菌感染不仅严重危害人类的健康,而且已经成为世界范围的棘手问题。因此,迫切需要挖掘作用于独特分子靶标的新型抗生素[1-2]。
2015年,美国药物学家Kim Lewis通过iChip(isolation chip)技术[3]在一株难培养的土壤微生物Eleftheria terrae中发现的新型抗生素teixobactin对多种革兰阳性致病菌,包括很多临床耐药菌具有显著的抑制活性,并且不易诱发耐药性[4]。Teixobactin能够分别和细菌细胞壁上肽聚糖前体脂质II和壁磷壁酸前体脂质III的保守序列结合,而这两个靶点是非常难通过突变而发展耐药性的[5]。这使teixobactin作为一种对抗耐药性致病菌的先导化合物显示出巨大的潜力[6-7]。本篇综述总结了自teixobactin发现以来的研究进展,重点介绍了其不易诱发耐药性的作用机制,并对其未来在生物合成方面的研究进行了展望。
1 Teixobactin的化学结构和生物活性
Teixobactin是由11个氨基酸残基组成的环缩肽,包含6个天然L-氨基酸,1个非天然氨基酸(L-alloenduracididine,L-allo-End10),N-Me-D-Phe和3个其他的D-氨基酸。C端的D-Thr8和L-Ile11通过酯键形成十三元环的四肽内酯亚结构(图1)。Teixobactin是细菌为了自我防御而产生的,必然经过了长期的自然进化和选择来形成最优化的结构,其显著的抑菌活性取决于这种独特的分子结构[8]。
1.1 L-allo-End—teixobactin中的非天然氨基酸
L-allo-End是一个含有五元结构环胍基团的非天然氨基酸。L-End是L-allo-End的非对映异构体,已被发现存在于多种天然产物中,比如甘露霉素(mannopeptimycin)[9]和恩拉霉素(enduracidins)[10],并对于维持其生物活性具有重要的作用。同位素标记实验证明L-End是由L-Arg产生的[11]。比较分析甘露霉素和恩拉霉素的生物合成基因簇,发现3对高度同源的酶:EndP/MppP(80%),EndQ/MppQ(68%)和EndR/MppR(75%)[12]。生化和结构研究表明,L-Arg经过PLP依赖的脱氨羟化酶MppP的催化生成2-羰基-4-羟基-5-胍基戊酸[13],后经脱羧酶MppR和PLP依赖的转氨酶MppQ催化生成L-End[14]。尽管L-allo-End和L-End在C4处有不同的立体化学构型,L-allo-End在teixobactin中的生物合成可能也遵循与L-End类似的途径。然而,在teixobactin的生物合成基因簇中并没有发现L-allo-End生物合成所需的同源基因。这可能是因为L-allo-End的生物合成基因簇位于E. terrae基因组的其他位置。这一假设还有待研究和考证。

图1 Teixobactin的化学结构Fig.1 Chemical structure of teixobactin.
L-allo-End的化学合成过程非常复杂。目前该氨基酸在市场上购买不到,因此成为了teixobactin化学合成的一大挑战。Rudolph课题组[15]首次报道了L-allo-End的化学合成,L-End作为一个副产物,与L-allo-End的产量比率是1:6。2015年,Craig课题组[16]报道了高度立体选择性地合成L-allo-End的新方法。从Boc-保护的反式-羟基脯氨酸中通过10步反应提供L-allo-End,具有超过50:1的非对映立体选择性。Payne课题组[17]报道了另一种方法来合成L-allo-End,以保护的L-Asp酯作为起始材料。在这个合成过程中,采用三仲丁基硼氢化锂的立体选择性酮还原反应来引入立体化学(dr: 5:1 (2S,4R):(2S,4S)),通过快速柱色谱除去次要非对映异构体产物。通过使用类似的策略,Reddy课题组[18]实现了L-allo-End的克级合成,足以用于teixobactin的化学全合成研究。
1.2 Teixobactin的生物活性
体外抑菌活性研究表明,teixobactin对大多数革兰阳性菌都具有显著的抑制活性,尤其是对梭状芽胞杆菌和炭疽杆菌的最低抑制浓度(minimal inhibitory concentration, MIC)达到了纳克级[4]。Teixobactin对临床上治疗周期长且成本高的结核杆菌也有很强的抑制活性[4]。对于多种耐药菌如MRSA和耐万古霉素肠球菌(VRE)等,teixobactin都具有显著的抑制活性,其MIC均在1μg/mL以下。Teixobactin不能有效抵抗大多数革兰阴性菌,但是对外膜通透性屏障缺陷的E. coliasmB1表现出良好的活性[4](表1)。体外毒理学实验研究表明,teixobactin没有明显的毒副作用[4]。

表1 Teixobactin对致病微生物的抑制活性[4]Tab. 1 Activity of teixobactin against pathogenic microorganisms[4]
体内活性研究显示,teixobactin在体内稳定存在,且毒性低。在小鼠MRSA感染败血症模型的研究中,teixobactin的半数保护量(PD50)为0.2mg/kg,低于临床治疗MRSA感染的万古霉素(2.75mg/kg)剂量[4]。Teixobactin的体内药动学结果也令人非常满意,小鼠接受20mg/kg的teixobactin,其血药浓度维持在MIC值以上的时间可达4h[4]。
2 Teixobactin的作用机制
大量实验研究表明,teixobactin不易诱发细菌产生耐药性。即使在4倍MIC剂量的teixobactin铺板时,也没能得到耐teixobactin的金黄色葡萄球菌和结核分枝杆菌[4]。在teixobactin的亚最低抑制浓度(sub-MIC)下连续传代27d,同样没有发现金黄色葡萄球菌的耐药性突变株[4]。因此,teixobactin作为对抗耐药性致病菌的先导化合物显示出巨大的潜力。这主要取决于teixobactin多靶标作用于细胞壁的特殊机制。
2.1 高度保守的肽聚糖前体—脂质II(LII)
脂质II(LII)是细菌细胞壁生物合成的最重要的中间产物之一。因其结构具有高度保守性,数十年来被公认为抗生素的理想靶标[19]。从结构上讲,一个LII分子由一个包埋在细胞膜中的细菌萜醇烃链(C55)、十一异戊烯基-N-乙酰氨基葡萄糖(UDPMurNAc)和N-乙酰胞壁酸(GlcNAc)的二糖、附着在MurNAc上的五肽(通常带有L-Ala-γ-D-Glu-L-DAP-DAla-D-Ala的序列)和连接细菌萜醇(C55)锚和MurNAc的焦磷酸盐基团(PP)组成。二糖和五肽形成的肽聚糖亚基进一步通过五肽相互交联,提供细胞壁的机械强度。该分子首先装配到细胞质侧的细菌萜醇(C55)锚上,然后易位至五肽连接的二糖所在的周质侧,从锚上释放出来构建细胞壁。剩余的C55-PP通过机制不明确的回收途径转移回细胞质侧。因此,阻止LII移位是通过干扰细胞壁合成途径中的关键步骤来杀灭细菌的一种策略。由于LII的高度保守性,细菌对于作用于LII的抗生素产生耐药性的几率非常低。
万古霉素和LII之间的相互作用已经被充分研究。万古霉素通过五肽的D-Ala-D-Ala片段上的5个氢键结合至LII,形成复合物[20]。这些复合物中的几个甚至可以通过D-Ala-D-Ala片段形成超级复合物[21]。然而,经过20年的进化和选择,细菌通过突变D-Ala-D-Ala的五肽末端为D-Ala-D-Lac从而对万古霉素产生了耐药性。万古霉素对突变的LII的结合亲和力降低了1000倍左右,因而大大降低了万古霉素的抑菌效果,产生耐药菌株[22]。尼生素(nisin)能有效抑制引起食品腐败的革兰阳性菌。它作用于LII中高度保守的焦磷酸基团(PP),目前没有发现耐药性[23]。马拉西汀(malacidins)是最近发现的可对抗多种耐药菌的钙依赖性抗生素,研究表明它也不易诱发细菌产生耐药性[24]。用马拉西汀处理金黄色葡萄球菌,细胞壁中积累UDP-MurNAc-五肽,说明马拉西汀作用的靶标也是LII。
2.2 壁磷壁酸(WTA)前体—脂质III(LIII)
磷壁酸是革兰阳性菌细胞壁所特有的成分。根据其在菌体上的分布,磷壁酸可分为2类,一类是壁磷壁酸,其末端以磷酸二酯键锚定到肽聚糖上;另一类是脂磷壁酸,锚定到细菌细胞膜上。抑制壁磷壁酸的生物合成对于细菌来说是致命性的,因为在细菌体内积累有毒中间体[25]。另外,壁磷壁酸可以固定自溶素,防止肽聚糖被水解。抑制壁磷壁酸生物合成有助于细菌释放自溶素,使细胞溶解,进而死亡[26]。
2.3 对抗耐药性—teixobactin独特的双重作用方式
Teixobactin可以双重靶向LII和LIII[4]。实验证明,teixobactin可以强效地抑制肽聚糖的生物合成,对于DNA、RNA和蛋白质均没有实质性作用[4]。添加纯化的LII可以阻止teixobactin抑制细菌生长的作用,说明teixobactin直接作用于LII[4]。化学计量分析表明teixobactin以2:1的比率结合LII[5]。进一步分析表明,teixobactin结合LII中高度保守的焦磷酸盐基团(PP)和MurNAc基团。并且teixobactin还能够有效地结合LIII中的UDP-PP-GlcNAc,有效地抑制壁磷壁酸的生物合成[4]。Teixobactin具有强有效的抑菌活性和无耐药性潜力,得益于它的双重作用方式,即同时抑制肽聚糖前体LII和壁磷壁酸前体LIII的生物合成(图2)。二者通过协同效应,导致细胞壁损伤,胞壁离域,蛋白酶自溶,随后细胞溶解[5]。这种高保守的双重靶标(LII和LIII)作用机制使致病菌几乎不可能对其产生耐药性。
最近,teixobactin-LII复合体的结构模型已经被阐明,发现了4种不同的结合模式(BM1、BM2、BM3和BM4)[27]。同时,结果表明,teixobactin的环状结构域对于识别LII具有重要作用[27]。这些新发现为进一步理解teixobactin与LII的作用机制提供了新思路。
3 Teixobactin及其类似物的化学合成
3.1 Teixobactin的化学全合成
成功地高效合成L-allo-End克服了teixobactin合成的主要障碍。目前为止,两个科研团队采用了固相多肽合成方法(SPPS),通过截然不同的合成途径完成了teixobactin的全合成[17,29]。
悉尼大学Payne课题组[17]利用Fmoc-SPPS首次实现了teixobactin的全合成。首先,他们先合成了可以直接安装入Fmoc-SPPS的经适当保护的L-allo-End,即FmocEnd(Cbz)2-OH;然后组装teixobactin的缩酚酞链;在弱酸性条件下裂解树脂,随后液相环化;然后在强酸性条件下,总体侧链去保护,包括L-allo-End的Cbz保护;最后产出teixobactin。通过24步过程,可以达到3.3%的总收率[17]。
香港大学李学臣课题组[29]报道了一种基于Ser/Thr连接的缩合反应来合成teixobactin。无保护肽段的N-Ser或N-Thr残基和另一个无保护肽段的C-水杨醛酯之间的化学选择性反应产生一种N, O-苄叉缩醛键合产物。它酸解时,在连接位置产生天然的肽键。含有N-Ser残基的环肽通过Fmoc-SPPS合成,含有C-水杨醛酯的线性肽片段通过Boc-SPPS合成,通过Ile6和Ser7的高效连接来合并线性的六肽和环化的五肽,从而合成teixobactin,缩合产物经HPLC纯化后收率为37%。值得注意的是,Arg10-teixobactin和Orn10-teixobactin的合成比teixobactin更顺利。这表明,天然形态的teixobactin的合成很可能会遇到一系列与类似物合成截然不同的挑战。
3.2 Teixobactin类似物的化学合成及其构效关系
L-Arg10-teixobactin是第一个报道的teixobactin类似物,报道时间比teixobactin的全合成还要早[30-31]。它常作为研究teixobactin构效关系的模型。目前,对于teixobactin构效关系的研究,全部基于相对容易合成的teixobactin类似物[8,30-43]。表2列举了目前成功合成的具有抑菌活性的teixobactin类似物。

图2 Lipid II和Lipid III的结构示意图以及teixobactin的作用靶点[28]Fig.2 Structures of Lipid II and Lipid III with the targets of teixobactin[28]
与天然的teixobactin相比,L-Arg10-teixobactin的抑菌活性有所降低,因此L-allo-End是维持teixobactin最佳生物活性的关键残基[30-31]。胍基对于维持活性是非必需的,因为Lys10-teixobactin和Orn10-teixobactin针对各种葡萄球菌的抑菌活性比Arg10-teixobactin更强,其MIC值在万古霉素(0.5μg/mL)范围内[32-33]。进一步的构效关系研究揭示了10位氨基酸残基的绝对立体化学不是必要的。而整个环状结构域的相对立体化学是非常关键的[32]。利用L-allo-End的电子等排体如Lys、Orn、L-2,4-Diaminobutyric acid (DAB)和L-1,3-diaminopropionic acid(DAP)[34]或疏水基团如Ile或Leu[35]L-allo-End10的类似物表现出出色的抑菌活性,证明L-allo-End或许并不是不可替代的。这一结论需要更进一步的体内外活性研究以及药效学研究。

表2 人工合成的teixobactin类似物及其抑菌活性Tab. 2 Synthesized teixobactin analogues with relatively good antibacterial activity
D-氨基酸对活性也起重要作用。当teixobactin结构中的4个D-氨基酸残基,即:NMe-D-Phe1、D-Gln4、D-allo-Ile5和D-Thr8全部或者逐个被L-氨基酸取代时,抑菌活性几乎全都丧失了[36-37]。Lys/Ala扫描(Lys/Ala scanning),即按顺序将每个残基替代为Lys/Ala,以研究疏水性与亲水性的平衡对于化合物保持抗菌活性的重要性。对Arg10-teixobactin进行Lys扫描[38]和对Lys10-teixobactin进行Ala扫描[39]的结果共同表明,疏水的Phe1、Ile2、Ile5、Ile6和Ile11对于保持活性非常重要,而极性不带电的Ser3、Gln4和中性的Ala9具有良好的耐受性。值得注意的是,Ala取代不带电的Ser7的类似物的抗菌活性显著下降,说明Ser7对于保持活性是至关重要的[39]。随后的晶体结构证明,Ala9的NH基团与Ser7的侧链形成了氢键[40]。进一步研究表明,维持teixobactin活性所需的最大正电荷数是3~4个,引入额外的正电荷将导致活性的完全损失[41]。另外,teixobactin的N-Me-Phe1对于保持活性非常重要,对其酰基化或烷基化都会导致活性完全丧失[42]。
Teixobactin类似物是否可以发展成为临床应用的新的抗菌药物,将化合物分子从发现阶段转换为临床使用的抗生素需要面对众多挑战,如针对广泛病原体高效的活性和无毒副作用之间的平衡,以及亲水性和疏水性之间的平衡以解决水溶性问题等。虽然Leu10-teixobactin和Ile10-teixobactin具有显著的抑菌活性,但是其疏水性的增加不利于其水溶性。将Ser3、Gln4和Ala9替换为阳离子的Arg残基可以模拟天然teixobactin的疏水性与亲水性之间的平衡。通过这种方式,带有两个阳离子残基的类似物,如D-Arg4-Leu10-Teixobactin实现了与teixobactin类似的疏水性和亲水性之间的最佳平衡,同时具有与teixobactin一致的抑菌活性[43]。体内毒理学研究和小鼠细菌性角膜炎模型研究均证明D-Arg4-Leu10-Teixobactin无细胞毒性作用[43]。这项工作在合成安全高效的简化的teixobactin类似物方面取得了重大进展,推动了新型对抗耐药性菌株的抗菌药物的研发进程。
4 Teixobactin的生物合成
生物信息学分析发现了teixobactin的生物合成基因簇(GenBank accession number KP006601),其包括两个NRPS编码基因,txo1和txo2。这两个NRPS编码基因共含有11个模块(module),计算机预测的每个腺苷酰结构域(A)的底物特异性都与teixobactin的结构相匹配[4]。因此,teixobactin遵循NRPS典型的共线性规则,其中每个模块负责整合新生肽链中的一个特定的氨基酸(图3)。通过L-Ile11与D-Thr8的内酯化反应将线性肽链从NRPS组装生产线上释放。甲基转移酶(MT)结构域存在于第1模块中,其负责D-Phe1的N-甲基化。值得注意的是,txo2包含两个串联硫酯酶(TE)结构域,这种结构在NRPS中是很罕见的。研究证明,两个串联硫酯酶在功能上是可互换的,并且很有可能协同作用,代表了一种前所未有的NRPS酶学卸载机制[44]。另一点值得提出的是,模块8包含一个额外的缩合(C)结构域,它的功能还有待研究。一些基因与txo1和txo2相邻,它们的功能尚不清楚,这些基因可能涉及调控、产物产出等。
近年来,本团队的RecET同源重组技术取得了重要进展,极大地促进了微生物生物合成基因簇组装、克隆和异源表达的研究[45-48]。这项技术是依托λ或Rac噬菌体的内源性DNA重组蛋白(Redα/β及RecE/T)而建立起来的一套完善的基因重组技术体系。其在大片段基因簇方面的有效应用,可以实现大片段、高重复的基因簇的载体化、工程化连接。近期,利用RecET同源重组技术,本团队完成了teixobactin生物合成基因簇(约54kb)的人工合成和组装,期望通过生物合成和异源表达的方式,更为高效、便利地大规模生产teixobactin及其类似物。
5 展望
自2015年teixobactin发现以来,科研学者们开展了大量的研究,包括作用机制、化学全合成、构效关系等。在此期间,先后使用和发展了多种高效的化学方法用于teixobactin及其类似物的合成,合成获得的化合物达上百种。显然,这些突破性成果是一个激动人心的开端。Teixobactin显著的抑菌活性、不易诱发耐药性的潜力及其对哺乳动物细胞的低毒作用等使其成为一种巨大开发前景的天然药物。
目前已经成功合成了与teixobactin抑菌活性一致的类似物,这项成果向合成安全高效的简化的teixobactin类似物的方向迈进了一大步,但是更进一步的构效关系研究以及毒理学和药理学研究也是必不可少的。另外,目前对于teixobactin与它的靶点之间的具体的作用方式还不是很清楚,这方面的继续研究对于理解teixobactin强有效的抑菌活性以及应对将来可能的耐药性发展意义重大。
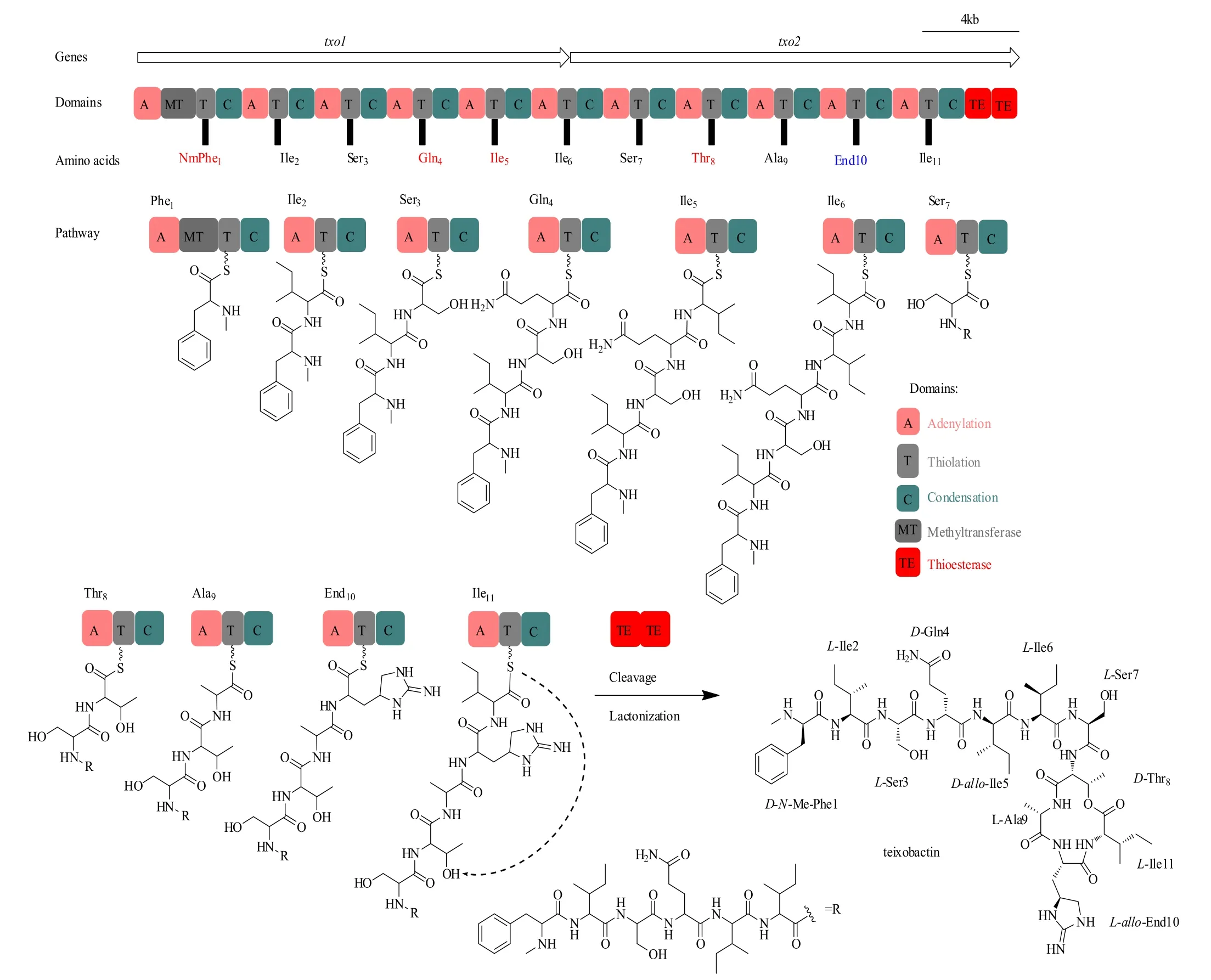
图3 预测的teixobactin生物合成途径[4]Fig.3 Predicted teixobactin biosynthetic pathway [4]
未来研究中另外一个重要的方面是teixobactin的生物合成,这在很大程度上滞后于化学合成研究。Teixobactin在野生菌中具体的生物合成途径以及后修饰是怎样的;L-allo-End在野生菌中的生物合成是如何实现的;如何提高teixobactin的总体产量,相信这方面的研究仍任重道远。RecET同源重组技术建立了对大型生物合成途径进行高通量的克隆、组装、修饰及异源表达的技术平台,首次实现了teixobactin基因簇的体外组装,为teixobactin基因组功能研究提供了极大的便利。
已知的天然产物仅仅代表自然界全部的天然产物生物合成能力的冰山一角,不可培养的微生物研究具有巨大的前景。大多数微生物有待挖掘,或者只是通过其基因组分析特征,至今还没有培养出来。随着微生物学和分子生物学技术的迅猛发展,挖掘这些尚未开发的新型抗生素对感染治疗具有重大意义。
猜你喜欢
临床肝胆病杂志(2022年6期)2022-11-25
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
中国计量大学学报(2022年2期)2022-07-18
肝博士(2022年3期)2022-06-30
西南农业学报(2021年10期)2021-12-14
食品安全导刊(2021年20期)2021-08-30
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
科技与创新(2020年12期)2020-11-28
中华养生保健(2020年3期)2020-11-16
