
高效液相色谱法测定原料药中溴芬酸钠及其杂质A含量
2018-11-20 07:42董莉,鲁军,窦佳
中国药业 2018年22期
董 莉,鲁 军,窦 佳
(辽宁省大连市药品检验所,辽宁 大连 116021)
溴芬酸钠的化学名称为2-氨基-3(-4-溴苯甲酰基)苯乙酸钠,是制备眼部炎症用药的原料。其结构与酮洛芬和双氯芬酸类似,能抑制环氧合酶介导的前列腺素类炎性介质的合成,具有强力消炎镇痛作用,作用强度为其他非甾体抗炎药的10倍[1-5]。其疗效和安全性均较好,特别对于白内障术后眼部发炎、疼痛及畏光症状效果显著[6-13]。本研究中建立的高效液相色谱(HPLC)法可测定原料药中溴芬酸钠及其杂质A的含量,且简便快速、专属性强、准确可靠,可有效控制该药品的质量[14-16]。现报道如下。
1 仪器与试药
1.1 仪器
Waters e2695型高效液相色谱仪,包括四元泵、自动进样器、柱温箱;Empower工作站,Waters 2489型紫外/可见光检测器,配备Waters 2998型二极管阵列检测器(Waters公司);PP-15E型酸度计,MSA 225s-100-DA型分析天平(德国Sartorius公司)。
1.2 试药
溴芬酸钠对照品(批号为WS/BRN/004-07)与溴芬酸钠杂质A对照品(批号为WS/BRW/003-07,纯度为96.8%),均由厂方提供;供试品为进口原料药(Indoco Remedies Ltd,批 号 分 别 为 RK14BRN003,RK14BRN004,RK14BRN001);甲醇为色谱纯,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件及系统适用性试验
色谱柱:HypersilBDSC18柱(250mm×4.6mm,5μm),流动相:甲醇-0.4%三乙胺溶液(用磷酸调pH至5.0)(55 ∶45);检测波长:266 nm;流速:1.0 mL /min;柱温:30℃;进样量:10 μL。在此条件下的色谱图见图1。
2.2 溶液制备
取本品20 mg,精密称定,置50 mL容量瓶中,加稀释剂适量,超声处理5 min,用稀释剂稀释至刻度,摇匀,作为供试品溶液。另取溴芬酸钠对照品适量,精密称定,加稀释剂制成每1 mL含0.4 mg的溶液,摇匀,作为溴芬酸钠对照品溶液。取杂质A对照品10 mg,置50 mL容量瓶中,加二氯甲烷1 mL使溶解,并用甲醇稀释至刻度,摇匀,即得对照品贮备液;精密量取对照品贮备液1 mL和供试品溶液0.5 mL,置同一50 mL容量瓶中,用稀释剂稀释至刻度,摇匀,精密量取1 mL,置10 mL容量瓶中,用稀释剂稀释至刻度,摇匀,作为溴芬酸钠杂质A对照品溶液。避光操作。
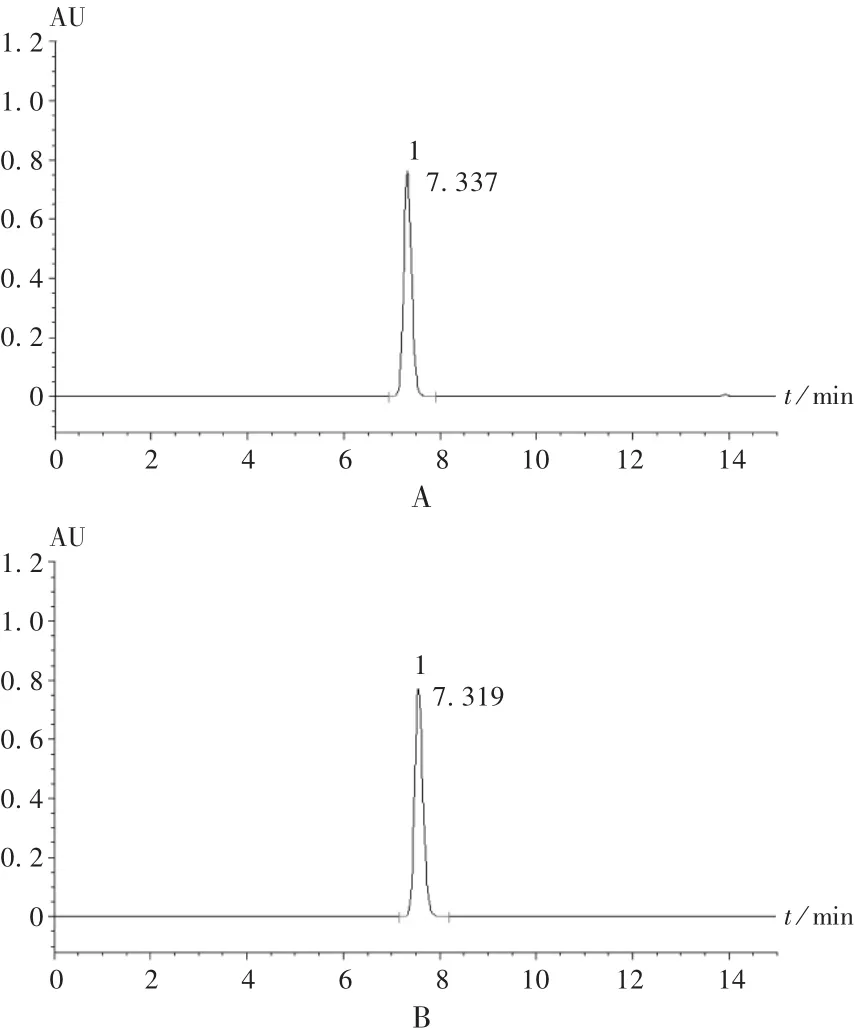
图1 高效液相色谱图
2.3 方法学考察
线性关系考察:称取溴芬酸钠对照品10,15,16,18,20,24,30 mg,精密称定,分别置 50 mL 容量瓶中,加稀释剂[甲醇-水(50∶50)]适量,超声使溶解,并稀释至刻度,摇匀,作为溴芬酸钠线性试验溶液。取溴芬酸钠杂质A[7-(4-溴苯甲酰基)-1,3-二氢-2H-吲哚 -2-酮]对照品10 mg,置50 mL容量瓶中,加二氯甲烷1 mL使溶解,并用甲醇稀释至刻度,摇匀,精密量取1 mL,置另一50 mL容量瓶中,用稀释剂稀释至刻度,摇匀,作为对照品贮备液;精密量取对照品贮备液5,7.5,10,15,20 mL,分别置 100 mL容量瓶中,用稀释剂稀释至刻度,摇匀,作为溴芬酸钠杂质A线性试验溶液。按2.1项下色谱条件分别进样2次,记录色谱图,以质量浓度(X)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程,溴芬酸钠为 Y=4.658×105X-1.328×105,r=0.9999(n=7);溴芬酸钠杂质 A 为 Y=2469X+2601,r=0.999 9(n=5)。结果表明,溴芬酸钠及其杂质 A质量浓度分别在 0.2 ~0.6 mg/mL 及 0.2 ~0.6 μg/mL范围内与峰面积线性关系良好。
稳定性试验:取室温避光放置 0,4,8,12,24 h 的供试品溶液,分别进样测定。结果峰面积未见明显变化,RSD为1.10%(n=5),表明该溶液避光放置日内稳定性良好,能满足测定要求。
精密度试验:取批号为RK14BRN003的样品,依法制备供试品溶液,连续进样5次。结果溴芬酸钠的平均含量为 99.99%,RSD 为 0.67%(n=5),表明仪器精密度良好。
重复性试验:取同一批(批号为RK14BRN003)样品5份,依法制备供试品溶液,进样测定。结果溴芬酸钠平均含量为 99.94%,RSD 为 0.44%(n=5),表明方法重复性好。
回收率试验:取线性关系考察项下的对照品溶液,按拟订色谱条件进样测定,每份样品各进样2次,测定,取平均值,计算回收率。结果见表1。

表1 回收率试验结果(n=7)
检出限和定量限确定:按2.1项下色谱条件,取线性关系考察项下溶液,稀释成质量浓度为 4,0.4,0.04,0.004 μg /mL 的溶液,分别取 10 μL,按拟订色谱条件进样分析。结果检测限按信噪比(S/N)=3∶1确定为1 ng/mL,定量限按 S/N=10∶1确定为4ng/mL。
2.4 样品含量测定
精密量取供试品溶液和溴芬酸钠对照品溶液各10 μL,按拟订色谱条件操作,用外标法计算含量。精密量取供试品溶液和溴芬酸钠杂质A对照品溶液各10 μL,按拟订色谱条件操作,记录色谱图至主成分峰保留时间的4倍,供试品溶液的色谱图中,如有与杂质A保留时间一致的峰,则按外标法以峰面积计算,不得超过 0.10。详见表 2及图 2。
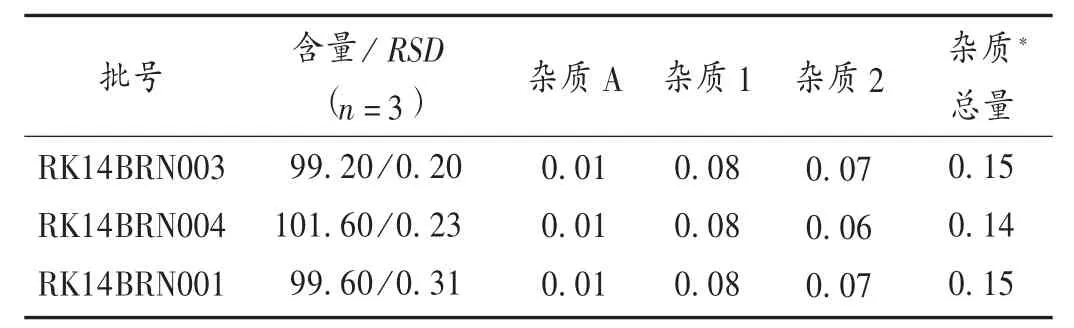
表2 样品含量测定结果(%)
2.5 强制降解试验
精密量取供试品溶液10 mL,分别精密加入30%过氧化氢溶液、稀盐酸、氢氧化钠溶液各1 mL,供试品溶液于60℃加热1 h后、相对湿度80%放置2 h,供试品溶液于日光照射2 h后,依法制备,即得破坏性试验a,b,c,d,e,f溶液,分别按拟订色谱条件测定。结果表明,该色谱条件可以将溴芬酸钠与各种条件下降解的有关物质分离。除80%相对湿度破坏影响不大外,其他温度、光照有些影响(降解产物质量约增加0.1%),氧化破坏和碱破坏影响较大,而酸破坏时溴芬酸钠几乎全部被破坏。详见图3。
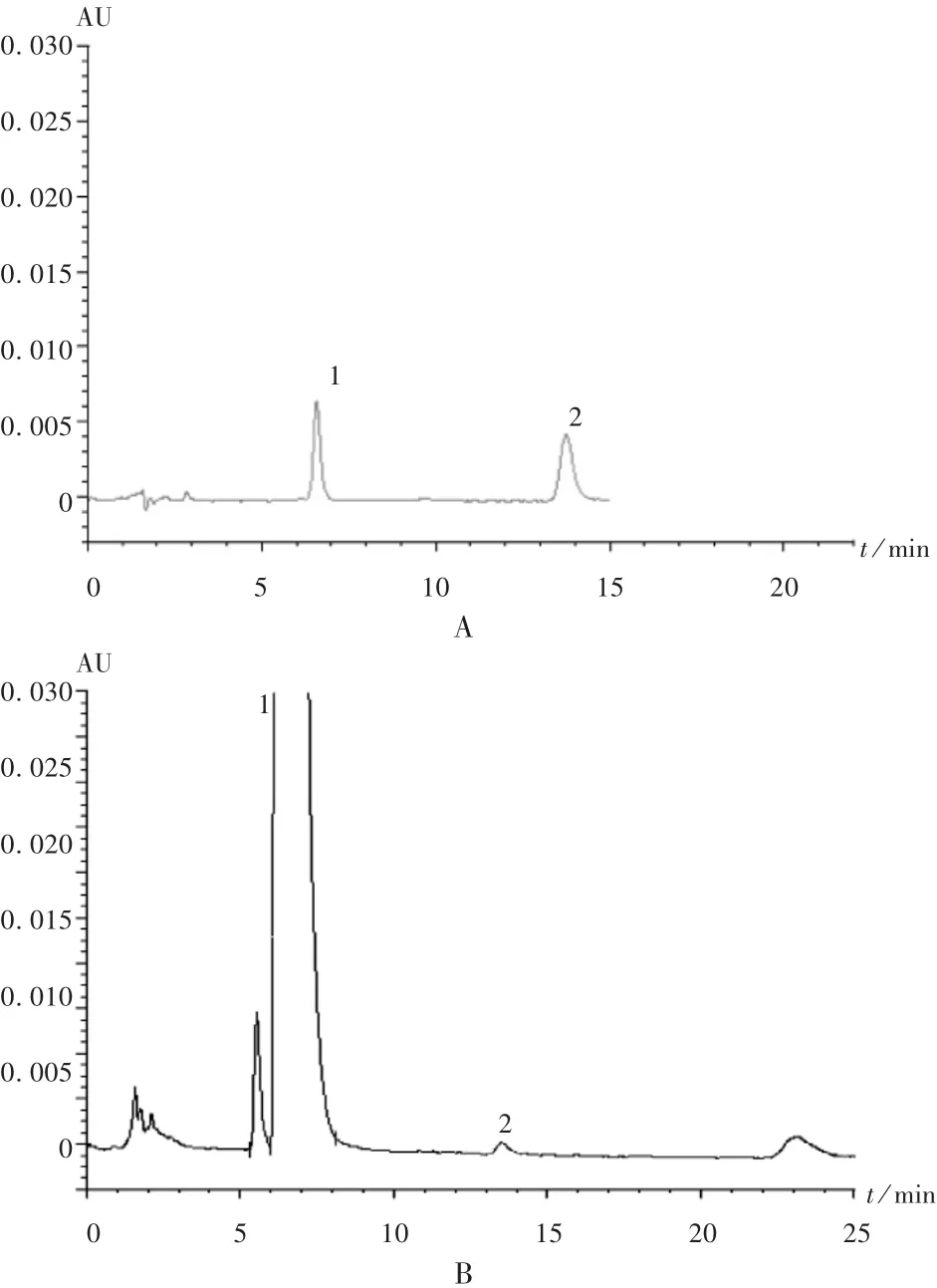
图2 含量测定高效液相色谱图
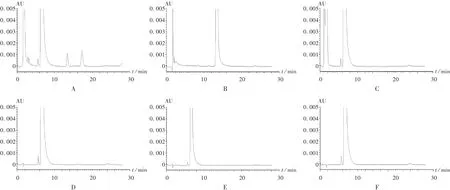
图3 强制降解试验高效液相色谱图
3 讨论
3.1 色谱柱耐受性考察
分别使用 Thermo BDS Hypersil C18柱(150 mm×4.6mm,5μm)、WatersSymmetryC18柱(250mm×4.6mm,5 μm)、Unitary C18柱(150 mm ×4.6 mm,5 μm)以及Kromasil C18柱(150 mm × 4.6 mm,5 μm)等色谱柱分析供试品溶液溴芬酸钠的杂质A与含量,其峰形及分离度均满意,故认为本方法对色谱柱的耐受性良好。
3.2 稳定性考察
经试验,该原料药不稳定,避光放置3~30 d,含量平均下降1% ~2%;对光比较敏感,光照3~30 d,有关物质总含量由0.2%增加到3.5% ~5.0%。供试品溶液颜色由黄色变为橘黄色。这其中溴芬酸钠杂质A的变化不大,而在相对保留时间0.9处及3.5处增加很多,故对其有关物质进行质量控制非常必要。
3.3 检测波长选择
以甲醇为溶剂,分别取溴芬酸钠对照品、溴芬酸钠杂质A对照品制成每1 mL含溴芬酸钠及溴芬酸钠杂质A10 μg的溶液,在200~400 nm波长范围内进行紫外扫描,结果溴芬酸钠及溴芬酸钠杂质A分别在266 nm及270 nm波长处有最大吸收峰。本研究中选择在266 nm波长处测定溴芬酸钠及杂质A的含量,效果满意。
3.4 杂质A考察
用本方法所测得的溴芬酸钠及溴芬酸钠杂质A的含量与梯度试验方法[3]比较,其结果完全一致。且有关物质多检出1个杂质,消除了梯度带来的基线波动造成的干扰,由样品测定和回收率测定结果可知,该方法准确性好,精密度高,重复性好,适用于测定溴芬酸钠原料及其中杂质A的含量。在加入盐酸强制降解试验中,溴被氯取代,且其保留时间与杂质A的保留时间一致。另外,在主峰相对保留时间0.9处均检出含量约0.08%的1个固定杂质,未能确定成分,有待进一步研究。
猜你喜欢
广州化工(2022年11期)2022-06-29
粘接(2021年5期)2021-06-29
中国应急管理科学(2021年4期)2021-04-13
森林工程(2020年5期)2020-09-17
核化学与放射化学(2020年4期)2020-08-21
中学生数理化·高一版(2020年9期)2020-01-02
学苑创造·A版(2019年9期)2019-11-07
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
学苑创造·B版(2017年1期)2017-02-21
