
意大利蜜蜂幼虫肠道发育过程中的差异表达 microRNA及其调控网络
2018-11-16 09:46郭睿杜宇熊翠玲郑燕珍付中民徐国钧王海朋陈华枝耿四海周丁丁石彩云赵红霞陈大福
中国农业科学 2018年21期
郭睿,杜宇,熊翠玲,郑燕珍,付中民,徐国钧,王海朋,陈华枝,耿四海,周丁丁,石彩云,赵红霞,陈大福
意大利蜜蜂幼虫肠道发育过程中的差异表达 microRNA及其调控网络
郭睿1,杜宇1,熊翠玲1,郑燕珍1,付中民1,徐国钧1,王海朋1,陈华枝1,耿四海1,周丁丁1,石彩云1,赵红霞2,陈大福1
(1福建农林大学蜂学学院,福州 350002;2广东省生物资源应用研究所,广州 510260)
【目的】微小RNA(microRNA,miRNA)是一类在转录后水平对mRNA进行负调控的关键调控因子。本研究旨在通过分析意大利蜜蜂(,简称意蜂)幼虫肠道发育过程中差异表达miRNA(differentially expressed miRNA,DEmiRNA)及其调控网络,提供miRNA的表达谱和差异表达信息,揭示DEmiRNA在幼虫肠道发育中的作用。【方法】利用small RNA-seq(sRNA-seq)技术对意蜂4、5和6日龄幼虫肠道样品(Am4、Am5和Am6)进行测序,将质控后的数据与西方蜜蜂()参考基因组进行比对,然后将比对上的序列标签(tags)注释到miRBase数据库,并利用TPM(tags per million)算法归一化处理所得miRNA的表达量,再通过相关生物信息学软件对miRNA进行表达量聚类、前体二级结构预测及差异表达分析。利用TargetFinder软件预测DEmiRNA的靶基因,使用Blast软件对靶基因进行GO和KEGG数据库注释,进而通过Cytoscape软件构建miRNA-mRNA的调控网络。采用茎环实时荧光定量PCR(Stem-loop RT-qPCR)验证测序数据的可靠性。【结果】意蜂幼虫肠道样品的测序分别得到10 841 644、12 037 678和9 230 496条有效序列标签;Am4 vs Am5比较组包含16个上调和10个下调miRNA,Am5 vs Am6比较组包含5个上调和7个下调miRNA。其中,novel-m0031-3p为两个比较组所共有,并结合5个与蜕皮激素诱导蛋白相关的靶基因;二者的特有DEmiRNA数分别为25和11个。Am4 vs Am5的26个DEmiRNA结合5 742个靶基因,其中2 725个靶基因可注释到GO数据库中的46个GO term,并主要富集在结合、细胞进程、代谢进程和单组织进程等;Am5 vs Am6的12个DEmiRNA结合3 733个靶基因,且其中2 725个靶基因富集在结合、细胞进程、单组织进程和代谢进程等41个GO term;此外,两个比较组中的DEmiRNA分别有1 046和676个靶基因可注释到116和92条KEGG代谢通路,且Am4 vs Am5比较组的DEmiRNA的靶基因富集在Wnt信号通路、Hippo信号通路、嘌呤代谢和内吞作用等通路上的数量均高于Am5 vs Am6比较组。进一步分析结果显示Am4 vs Am5中的上调和下调miRNA可分别结合611和85个靶基因,其中ame-miR-6052结合的靶基因数最多,可通过结合5个靶基因,参与对细胞色素P450的调控;miR-281-x结合49个靶基因,并间接调控组氨酸代谢、TGF-信号通路以及Hippo信号通路;Am5 vs Am6中的上调和下调miRNA可分别结合43和431个靶基因,其中miR-iab-4-x结合靶基因数量最多,并广泛参与调控背腹轴的形成、Hippo信号通路、Wnt信号通路、FoxO信号通路、Notch信号通路以及mTOR信号通路等与生长发育相关的通路。调控网络分析结果表明,DEmiRNA与靶基因间形成较为复杂的调控网络,DEmiRNA居于调控网络的中心位置,而mRNA位于调控网络的外周。最后,通过茎环实时荧光定量PCR对随机选取的3个DEmiRNA进行验证,结果证实了测序数据的可靠性。【结论】在全基因组水平对意蜂幼虫肠道的DEmiRNA及其靶基因进行预测和分析,并对DEmiRNA的调控网络进行构建及分析,发现意蜂幼虫通过调节包括ame-miR-6052、miR-iab-4-x、miR-281-x和novel-m0031-3p在内的多个miRNA的表达水平对肠道生长和发育进行调控,研究结果不仅提供了意蜂肠道发育过程的miRNA表达谱及差异表达信息,也为阐明意蜂幼虫肠道的发育机理打下基础。
意大利蜜蜂;幼虫肠道;发育;微小RNA;靶基因;调控网络
0 引言
【研究意义】微小RNA(microRNA,miRNA)是一类长度约为18—25个核苷酸的非编码RNA(non-coding RNA,ncRNA),通过对mRNA的负调控作用而广泛参与动物[1-2]、植物[3-4]和微生物[5-6]的各类生物学过程。意大利蜜蜂(,简称意蜂)是我国养蜂生产中的主要蜂种,在经济创收和生态维持等方面具有重要价值。利用small RNA-seq(sRNA-seq)技术对意蜂4、5和6日龄幼虫肠道进行测序,通过生物信息学分析方法对肠道发育过程中的差异表达miRNA(differentially expressed miRNA,DEmiRNA)进行全面分析,并分别构建上调和下调miRNA的调控网络,对揭示意蜂幼虫肠道发育过程中miRNA的差异表达规律、肠道发育相关关键miRNA的筛选和功能研究以及阐明肠道发育的机理具有重要意义。【前人研究进展】西方蜜蜂()基因组[7]的公布为其组学和分子生物学研究奠定了基础。此后,随着高通量测序技术、生物信息学分析方法和软件的革新、融合及应用,蜜蜂的转录组学研究取得了较大进展[8-9]。对蜜蜂的miRNA开展了一些研究,如Ashby等[10]研究发现,miR-184、miR-bantam和miR315参与了蜜蜂的细胞分化、组织结构重塑以及级型分化;陈晓[11]对蜂王卵巢激活、产卵抑制和产卵恢复过程中的DEmiRNA进行靶基因预测和分析,发现多个DEmiRNA与卵巢激活和产卵密切相关;石元元[12]研究发现,ame-bantam、ame-let-7和ame-miR-8参与了东方蜜蜂()4日龄蜂王幼虫和工蜂幼虫的Wnt信号通路,并与雌性蜜蜂级型分化的分子机理密切相关。此外,蜜蜂肠道不仅是食物消化、营养吸收和利用的主要场所,也是抵御病原入侵的重要免疫器官。前人对蜜蜂肠道的研究主要集中在肠道菌群的群落结构和功能预测方面[13-14],但有关蜜蜂及其幼虫肠道发育的研究极为滞后,肠道发育的分子机理仍不明确。笔者所在课题组前期已在mRNA组学水平对意蜂幼虫肠道进行了全面的转录组学研究,通过差异表达基因(DEG)和趋势分析揭示了意蜂幼虫肠道发育过程中的基因表达谱和差异表达规律[15],意蜂幼虫肠道响应球囊菌()胁迫的免疫应答[16],以及意蜂幼虫与球囊菌之间的互作[17]。【本研究切入点】目前为止,意蜂幼虫肠道发育过程中的miRNA表达谱仍然缺失,相关miRNA在幼虫肠道发育过程中的作用还不明确。【拟解决的关键问题】结合sRNA-seq技术对意蜂4、5和6日龄幼虫肠道样品进行测序和分析,通过DEmiRNA及其靶基因的预测和分析、调控网络的构建和分析,提供意蜂幼虫肠道发育过程中的miRNA表达谱和差异表达信息,揭示DEmiRNA在肠道发育中的作用。
1 材料与方法
试验于2017年9月至2018年5月在福建农林大学蜂学学院蜜蜂保护实验室进行。
1.1 生物材料
供试意蜂幼虫取自福建农林大学蜂学学院教学蜂场。
1.2 意蜂幼虫人工饲养及样品测序
按照王倩等[18]的方法配制幼虫饲料,从健康意蜂蜂群中提取巢脾,将2日龄幼虫移至已预置50 μL饲料的24孔细胞培养板中,在35℃,相对湿度(RH)为90%的培养箱中饲养,每隔24 h更换饲料。分别剖取4、5和6日龄意蜂幼虫肠道(分别记为Am4、Am5和Am6),装入EP管后放入液氮速冻,随后保存在-80℃的超低温冰箱。进行3次生物学重复。Am4的3个生物学重复分别为Am4-1、Am4-2和Am4-3,Am5的3个生物学重复分别为Am5-1、Am5-2和Am5-3,Am6的3个生物学重复分别为Am6-1、Am6-2和Am6-3。上述9个肠道样品委托广州基迪奥生物科技有限公司对进行单端测序,测序平台为Illumina MiSeq。原始数据已上传NCBI SRA数据库,BioProject号:PRJNA408312。
1.3 测序数据的质控及定位
对原始下机数据,利用Perl脚本剔除衔接子(adaptor)、未知碱基N和低质量的reads,从而得到高质量的序列(clean reads),保证后续数据分析的准确性。通过Bowite软件将各样品的tags序列比对到GenBank及Rfam(11.0)数据库,过滤核糖体RNA(rRNA)、胞质小RNA(scRNA)以及核内小RNA(snoRNA)等ncRNA和重复序列,得到miRNA的非注释标签序列(unannotated tags)。继而利用Bowit 软件将非注释标签序列与西方蜜蜂的参考基因组(assembly Amel 4.5)的序列进行比对,得到相关tags在参考基因组上的位置信息,即为mapped tags。
1.4 DEmiRNA的预测
利用miRDeep2软件[19]将mapped tags与miRBase数据库中已知miRNA前体序列进行比对,从而鉴定已知miRNA的表达,同时得到可能的前体序列。对各样本中miRNA进行表达量的统计,并通过TPM(tags per million)算法公式(TPM=T×106/N,T表示miRNA的tags,N表示总miRNA的tags)对全部miRNA的表达量进行归一化处理。利用R软件计算各样品之间的相关性系数。DEmiRNA(Am4 vs Am5、Am5 vs Am6和Am4 vs Am6)的筛选标准为-value≤0.05且|log2Fold change|≥1。
1.5 DEmiRNA靶基因预测及分析
根据DEmiRNA与对应物种的基因序列信息,利用TargetFinder软件[20]进行靶基因预测,并利用Blast软件将预测靶基因序列与GO(Gene Ontology)、KEGG数据库比对,获得靶基因的注释信息。利用RNAhybrid、TargetScan和Miranda软件预测DEmiRNA结合的靶基因,根据上述靶向结合关系构建miRNA-mRNA的调控网络并通过Cytoscape软件[21]将其可视化。
1.6 DEmiRNA的茎环实时荧光定量PCR(Stem-loop RT-qPCR)验证
为了验证sRNA-seq数据的可靠性,随机选取3个DEmiRNA(miR-7964-y、miR-8516-x和miR-3747-x)进行Stem-loop RT-qPCR验证。根据所选DEmiRNA的序列,参照Chen等[22]的方法,利用DNAMAN软件(Lynnon Biosoft公司,美国)设计特异性的Stem-loop引物、上游引物和下游引物,委托上海生工生物工程有限公司进行引物合成。选择snRNA U6作为内参。利用RNA抽提试剂盒(Axygen公司,美国)分别提取Am4、Am5和Am6的总RNA,利用Stem-loop引物进行反转录得到相应的cDNA,作为模板进行qPCR。反应体系(20 μL)中含有SYBR Green Dye 10 μL,上下游引物各1 μL,cDNA模板DNA 1 μL,Rox 0.44 μL,DEPC水补至20 μL。在ABI 7500荧光定量PCR仪(ABI公司,美国)中进行反应,反应条件:95℃预变性1 min,95℃变性15 s,48℃延伸30 s,共40个循环,最后72℃延伸45 s。所选miRNA的相对表达量采用2-△△Ct法计算。每个反应进行3个生物学重复和3次平行重复。
2 结果
2.1 数据质控与评估
本研究中,3个幼虫肠道样品的sRNA-seq分别产生13 186 921、14 255 967和10 921 897条clean reads,经严格过滤后得到的clean tags数分别为10 841 644(82.22%)、12 037 678(84.44%)和9 230 496(84.51%)条(表1)。Am4、Am5和Am6组内各生物学重复之间的Pearson相关系数均在0.9734以上,说明各样品的重复性较好(图1)。上述结果说明本研究的测序数据质量良好,可用于进一步分析。
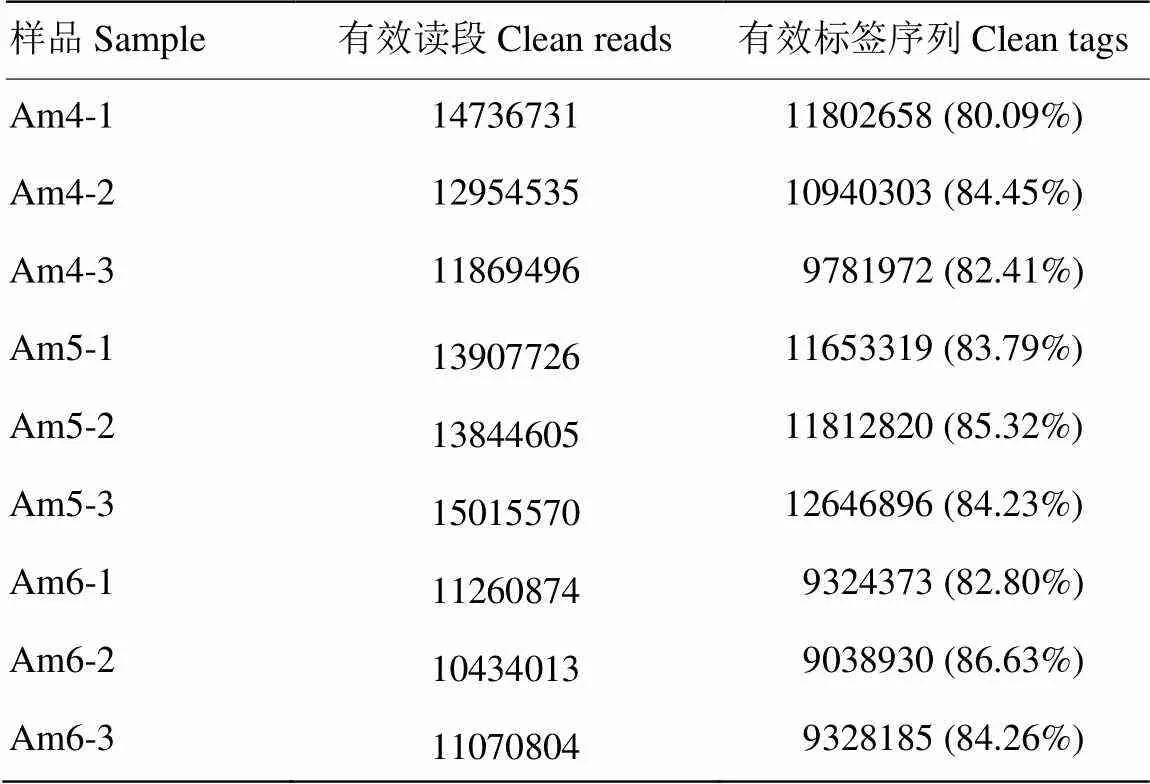
表1 sRNA-seq数据统计
2.2 意蜂幼虫肠道的DEmiRNA分析
Am4 vs Am5比较组中有26个miRNA差异表达,包括16个上调miRNA和10个下调miRNA。Am5 vs Am6 比较组共有12个DEmiRNA,包括5个上调miRNA和7个下调miRNA。Am 4 vs Am6比较组中有41个miRNA差异表达,包括15个上调miRNA和26个下调miRNA。分别对Am4 vs Am5和Am5 vs Am6的DEmiRNA进行表达量聚类分析,结果显示2个比较组中DEmiRNA的变化倍数差异明显,多数DEmiRNA的差异表达幅度较小(图2-A、2-B)。进一步分析发现,Am4 vs Am5和Am5 vs Am6中DEmiRNA分别包含3个和2个novel miRNA,其中novel-m0031-3p为2个比较组所共有。利用miRDeep2软件对novel miRNA的前体二级结构进行预测,结果显示它们均有标志性的茎环结构(图2-C—2-F)。
2.3 意蜂幼虫肠道的DEmiRNA的靶基因预测及功能注释
利用TargetFinder软件对意蜂幼虫肠道的DEmiRNA进行靶基因预测,Am4 vs Am5、Am5 vs Am6的DEmiRNA可分别预测出5 742和3 733个靶基因。对上述靶基因进行GO数据库注释,结果显示Am4 vs Am5的DEmiRNA的2 725个靶基因共涉及46个GO term,富集基因数最多的是结合(1 612 gene)、细胞进程(1 537 gene)、代谢进程(1 232 gene)、单组织进程(1 197 gene)、催化活性(1 020 gene)、细胞膜(761 gene)、细胞(757 gene)、细胞组件(757 gene)、细胞膜组件(745 gene)、细胞器(578 gene)等(图3);Am5 vs Am6的DEmiRNA的1 785个靶基因可注释到41个GO term,富集基因数最多的是结合(1 068 gene)、细胞进程(1 026 gene)、单组织进程(847 gene)、代谢进程(799 gene)、催化活性(591 gene)、细胞膜(533 gene)、细胞膜组件(529 gene)、细胞(448 gene)、细胞组件(448 gene)、生物学调控(348 gene)等(图3)。说明随着发育时间的延长,意蜂幼虫肠道的DEmiRNA数、靶基因数及其涉及的GO term数逐渐减少;DEmiRNA广泛参与意蜂幼虫肠道的新陈代谢、细胞生命活动及免疫防御。

图1 各意蜂幼虫肠道样品的不同生物学重复间的Pearson相关性
进一步对DEmiRNA的靶基因进行KEGG代谢通路(pathway)富集分析,结果显示Am4 vs Am5中的1 046个DEmiRNA的靶基因可注释到116条pathway,其中富集基因数最多的是Wnt信号通路(140 gene)、嘌呤代谢(87 gene)、Hippo信号通路(80 gene)、内吞作用(64 gene)、光传导(60 gene)、神经活性配体-受体相互作用(57 gene)、FoxO信号通路(43 gene)、内质网中蛋白质的加工(41 gene)、磷脂酰肌醇信号系统(38 gene)、泛素介导的蛋白水解(36 gene)等(图4-A),说明相应的miRNA参与到意蜂4和5日龄幼虫肠道发育过程中的新陈代谢、蛋白质合成、免疫防御以及相关信号通路的调控。
Am5 vs Am6中的676个DEmiRNA的靶基注释到92条pathway,并主要富集在Wnt信号通路(109 gene)、光传导(69 gene)、Hippo信号通路(66 gene)、神经活性配体-受体相互作用(57 gene)、嘌呤代谢(42 gene)、内吞作用(38 gene)、背腹轴形成(30 gene)、mRNA监视(28 gene)、Hedgehog信号通路、昼夜节律(24 gene)等(图4-B),说明相应的miRNA同样广泛参与了意蜂幼虫5和6日龄肠道的生长发育过程中各类新陈代谢以及信号通路的调控过程。
2.4 意蜂幼虫肠道DEmiRNA的调控网络分析
利用软件预测DEmiRNA结合的靶基因并通过Cytoscape软件进行可视化,结果显示Am4 vs Am5中上调miRNA可结合611个靶基因,下调miRNA可结合85个靶基因,上调(或下调)miRNA与靶基因形成较为复杂的调控网络,DEmiRNA居于调控网络的中心位置,而靶基因处于调控网络的外周;其中,所有DEmiRNA均可连接2个靶基因以上,ame-miR- 6052结合的靶基因数多达204个(图5-A、5-B)。Am5 vs Am6中上调和下调miRNA可分别结合43和431个靶基因,上调(或下调)miRNA同样与靶基因形成复杂的调控网络,所有DEmiRNA均可连接3个以上的靶基因,其中miR-iab-4-x结合的靶基因数最多,达到125个(图5-C、5-D)。
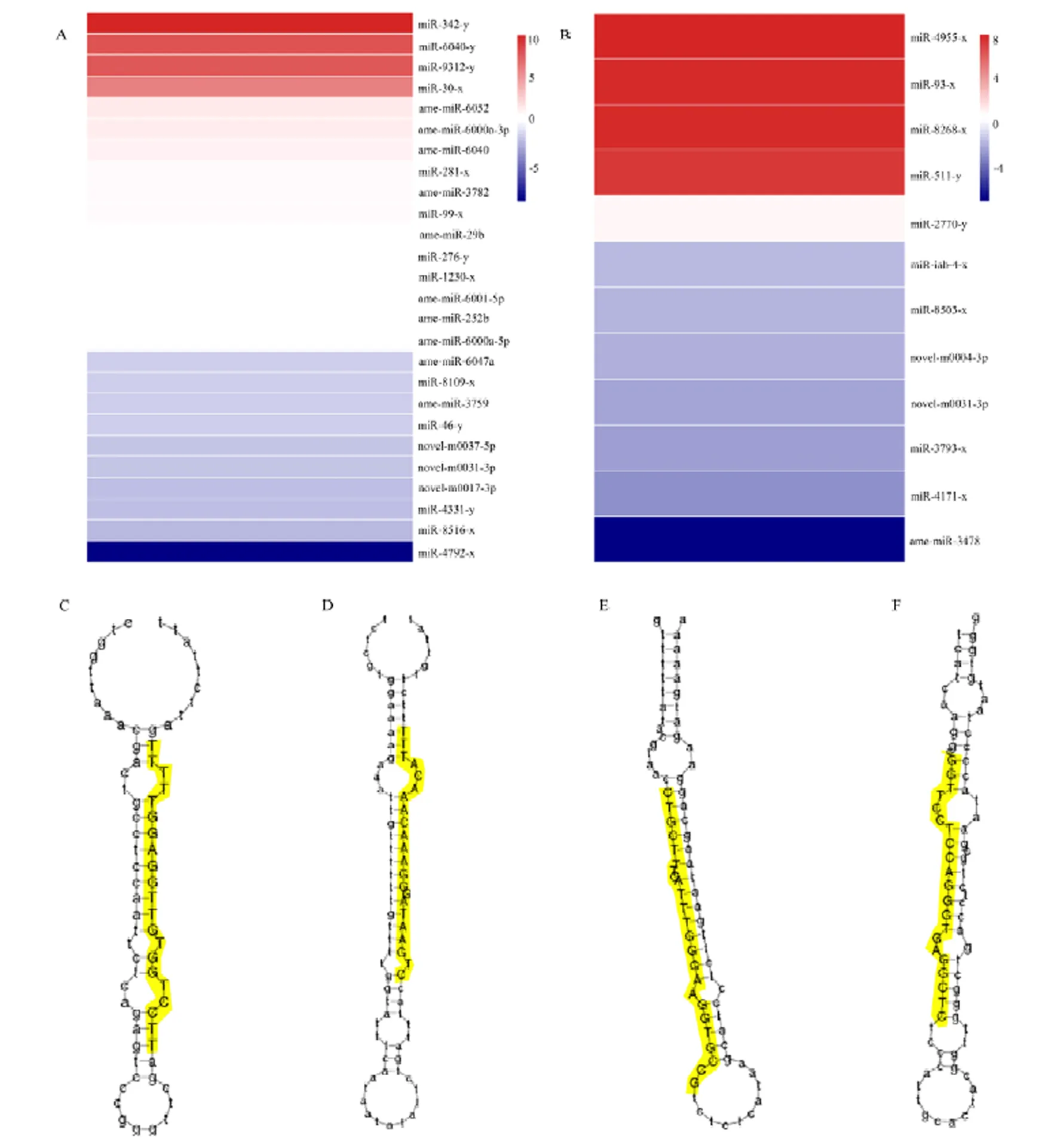
A:Am4 vs Am5中DEmiRNA的表达量聚类Expression clustering of DEmiRNA in Am4 vs Am5;B:Am5 vs Am6中DEmiRNA的表达量聚类Expression clustering of DEmiRNA in Am5 vs Am6;C:novel-m0004-3p前体的二级结构Secondary structure of precursor of novel-m0004-3p;D:novel-m0017-3p前体的二级结构Secondary structure of precursor of novel-m0017-3p;E:novel-m0031-3p前体的二级结构Secondary structure of precursor of novel-m0031-3p;F:novel-m0037-5p前体的二级结构Secondary structure of precursor of novel-m0037-5p。黄色区域为novel miRNA的成熟序列Yellow regions indicate mature sequences of novel miRNA

图3 意蜂幼虫肠道DEmiRNA的靶基因的GO数据库注释
2.5 意蜂幼虫肠道DEmiRNA的RT-qPCR验证
随机挑取3个DEmiRNA(miR-7964-y、miR-8516-x和miR-3747-x)进行RT-qPCR验证,结果显示它们的表达水平的变化趋势和测序数据中相应DEmiRNA的表达水平的变化趋势一致(图6),说明本研究中的测序数据真实可靠。

A:Am4 vs Am5中的DEmiRNA的靶基因Target genes of DEmiRNA in Am4 vs Am5;B:Am5 vs Am6中的DEmiRNA的靶基因Target genes of DEmiRNA in Am5 vs Am6

A:Am4 vs Am5中上调miRNA的miRNA-mRNA网络miRNA-mRNA networks of up-regulated miRNA in Am4 vs Am5;B:Am4 vs Am5中下调miRNA的miRNA-mRNA网络miRNA-mRNA networks of down-regulated miRNA in Am4 vs Am5;C:Am5 vs Am6中上调miRNA的miRNA-mRNA网络miRNA-mRNA networks of up-regulated miRNA in Am5 vs Am6;D:Am5 vs Am6中下调miRNA的miRNA-mRNA网络miRNA-mRNA networks of down-regulated miRNA in Am5 vs Am6
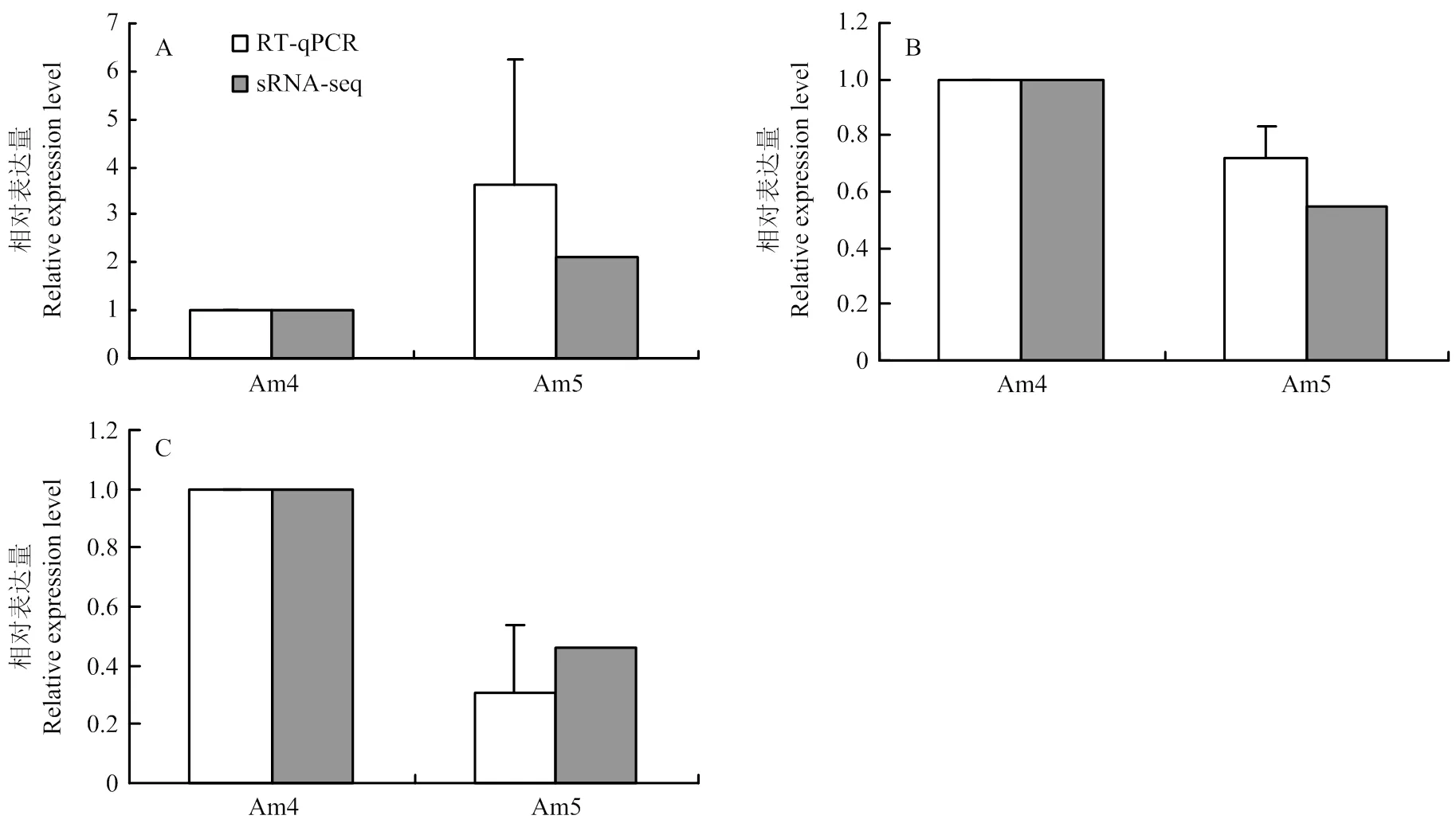
A:miR-7964-y;B:miR-8516-x;C:miR-3747-x
3 讨论
MiRNA作为一种重要的基因表达调控因子,在昆虫的各种生物学进程发挥关键作用[23]。家蚕()和小菜蛾()不同发育阶段和不同组织中的miRNA表达谱研究揭示了miRNA在其生长发育[24]、变态[25]及行为反应[26]等过程中的重要调控功能。相比于果蝇、家蚕等模式昆虫,蜜蜂的miRNA研究相对滞后。陈璇等[27-28]曾对三型蜂不同发育阶段的混合小RNA文库进行了测序,发现了267个novel miRNA,并进一步对蜂王和工蜂4和5日龄幼虫的DEmiRNA及其靶基因进行了分析,为蜜蜂发育和级型分化关键时期的miRNA调控网络研究打下了基础。目前,有关蜜蜂幼虫肠道发育过程miRNA表达谱的研究十分滞后,miRNA在肠道发育中的调控机理仍不明确。本研究利用sRNA-seq技术对意蜂4、5和6日龄幼虫肠道进行测序,通过生物信息学方法对邻近日龄幼虫肠道的miRNA进行差异表达分析,分别预测出26和12个DEmiRNA,说明DEmiRNA的数量在意蜂幼虫肠道发育过程中有逐渐减少的趋势;进一步分析发现Am4 vs Am5和Am5 vs Am6中的特有DEmiRNA数分别为25(miR-342-y、ame-miR-3759和miR-4331-y等)和11个(miR-4955-x、miR-46-y和ame-miR-3478等),二者包含1个共有DEmiRNA(novel-m0031-3p),且novel-m0031-3p的表达水平随日龄的增加呈逐渐下调趋势,推测其通过下调表达量减少对靶基因的抑制作用,从而在意蜂幼虫肠道的不同发育阶段均发挥基础性的调控作用,而特有DEmiRNA在幼虫肠道的不同发育阶段发挥特殊的调控功能。
蜜蜂幼虫肠道内存在简单的共生菌,为尽量减小意蜂幼虫肠道内共生菌对测序数据的影响,本研究一方面在人工饲喂过程中尽量减少环境微生物的带入,例如对所用器具进行高压蒸汽灭菌,用75%酒精对人工气候箱进行擦洗消毒,更换饲料时在酒精灯旁操作等;另一方面通过对原始数据进行严格的过滤和质控、比对西方蜜蜂基因组以消除肠道共生菌数据的影响,昆虫和微生物的物种亲缘关系较远,二者的基因保守性很低,因而比对西方蜜蜂基因组的数据理论上应为意蜂幼虫肠道本身的数据。
蜜蜂肠道不仅是食物消化、营养吸收和利用的主要场所,也是抵御病原入侵的重要免疫器官。本研究发现,Am5 vs Am6中的12个DEmiRNA的靶基因富集在了各类能量和物质代谢通路,如氮代谢(1 gene)、氨基酸代谢(32 gene)、碳水化合物代谢(69 gene)和脂质代谢(74 gene)等,表明相应的DEmiRNA参与了意蜂幼虫肠道能量和物质代谢的调控;还发现部分靶基因注释到与生长、发育相关的代谢通路和信号通路,包括背腹轴形成(30 gene)、Hippo信号通路(66 gene)、Wnt信号通路(109 gene)等,表明相应的DEmiRNA在生长和发育过程中发挥重要的调控作用。此外,分别有38和13个靶基因注释到内吞作用和泛素介导的蛋白质水解等细胞免疫通路,分别有6和4个靶基因注释到MAPK和Jak-STAT等体液免疫通路,表明相应的DEmiRNA参与了意蜂幼虫肠道的免疫防御过程。本研究中,Am4 vs Am5中的26个DEmiRNA的靶基因同样也广泛参与生长发育、新陈代谢以及免疫防御相关的代谢通路。Am4 vs Am5中的DEmiRNA及其靶基因数均高于Am5 vs Am6,意蜂5日龄幼虫体积近乎4日龄幼虫的2倍,但6日龄幼虫体积稍大于5日龄幼虫,推测意蜂4—5日龄幼虫的发育时期需要更多的DEmiRNA参与到生长发育、新陈代谢等方面的调控。
蜕皮激素(ecdysteroid,Ec)是昆虫体内的重要激素之一,参与昆虫生长、变态和生殖的整个生命活动,其滴度的阶段性增加是昆虫生长和发育的必要条件[29]。novel-m0031-3p在Am4 vs Am5和Am5 vs Am6中均有表达,且其结合的5个靶基因(XM_006564318.2、XM_006564319.2、XM_006564320.2、XM_006564321.2和XM_016914663.1)均涉及Ec诱导蛋白的调控,有研究表明蜕皮触发激素基因可在家蚕幼虫蜕皮和变态发育过程中起到关键调控作用[30],因此推测novel-m0031-3p通过调控Ec滴度使其达到动态平衡,从而保证意蜂幼虫肠道的正常发育。miR-281是一种高度保守的miRNA,在家蚕幼虫阶段中呈高量表达,与神经发育、组织生长密切相关[25]。周艳河[31]研究发现,受到高剂量登革病毒侵染的白纹伊蚊()中肠内miR-281会靶向调控自身基因从而影响病毒的复制水平;熊慧萍[32]对位于可变内含子区的黑腹果蝇()的miR-281-1/2基因转录和启动子进行分析,发现miR-281只在3龄幼虫、蛹和成虫期表达,且在不同发育阶段miR-281转录起始位点的转录效率存在差异。本研究中,miR-281-x是Am4 vs Am5中的特有DEmiRNA,其结合的49个靶基因涉及TGF-信号通路(8 gene)、Hippo信号通路(7 gene)、背腹轴的形成(3 gene)和组氨酸代谢(3 gene)等代谢通路,推测其参与对意蜂幼虫肠道早期生长和发育的调控,但miR-281-x的时空表达谱需进一步研究。miR-iab-4可通过调控Hox编码蛋白的基因表达间接影响黑腹果蝇的翅形成过程,miR-iab-4的突变会影响果蝇幼虫的自身调节能力[33]。本研究中,miR-iab-4-x为Am5 vs Am6中的特有DEmiRNA,其结合的125个靶基因涉及Hippo信号通路(14 gene)、Wnt信号通路(9 gene)、FoxO信号通路(8 gene)、Notch信号通路(7 gene)、mTOR信号通路(6 gene)和背腹轴的形成(5 gene)等与生长发育相关的代谢通路,推测其参与意蜂幼虫肠道发育后期的调控,miR-iab-4-x的下调表达可能对意蜂幼虫肠道发育过程发挥重要作用。
一个miRNA可以同时靶向调控多个mRNA,反之亦然。为进一步揭示DEmiRNA的作用,本研究通过序列匹配关系预测DEmiRNA结合的靶基因,并构建了二者的调控网络(图5),发现部分上调和下调的miRNA位于调控网络的中心位置且结合较多的mRNA,如ame-miR-6052与miR-iab-4-x可分别结合204和125个靶基因,具有很高的连通性,表明二者可能在意蜂幼虫肠道的生长和发育过程中发挥关键的调控功能。细胞色素P450作为一种重要的解毒酶,广泛存在于昆虫的脂肪体、马氏管和中肠内,且在中肠的含量最高;其也可参与内源性物质代谢,在生物体内发挥重要的作用[34-35]。本研究发现Am4 vs Am5中特有的ame-miR-6052表达量上调,其结合的5个靶基因(XM_006559340.2、XM_006559341.1、XM_016912202.1、XM_016916903.1和XM_623618.5)与细胞色素P450的调节有关,推测ame-miR-6052可参与意蜂幼虫肠道对异源物质的降解、新陈代谢过程的调控,下一步可通过合成miRNA mimic、miRNA inhibitor对其进行过表达和敲减,从而深入探究其功能。
4 结论
结合高通量测序技术和生物信息学分析方法对意蜂幼虫肠道的DEmiRNA进行了全基因组水平的预测和分析,并对DEmiRNA-mRNA调控网络进行构建及分析,发现意蜂幼虫通过调节包括ame-miR-6052、miR-iab-4-x、miR-281-x和novel-m0031-3p在内的多个miRNA的表达水平对肠道生长和发育进行调控,研究结果不仅提供了意蜂肠道发育过程的miRNA表达谱及差异表达信息,也为阐明意蜂幼虫肠道的发育机理打下了基础。
[1] Azzam G, Smibert P, Lai E C, LIU J L.Argonaute 1 and its miRNA biogenesis partners are required for oocyte formation and germline cell division., 2012, 365(2): 384-394.
[2] Asgari S. MicroRNA functions in insects., 2013, 43(4): 388-397.
[3] Xu L, Hu Y G, Cao Y, LI J R, MA L G, LI Y, QI Y J. An expression atlas of miRNAs in., 2018, 61(2): 178-189.
[4] Liu M, Yu H, Zhao G, HUANG Q, LU Y, OUYANG B. Profiling of drought-responsive microRNA and mRNA in tomato using high-throughput sequencing., 2017, 18(1): 481.
[5] Yu Z Q, Gao X L, Liu C N, LV X P, ZHENG S M. Analysis of microRNA expression profile in specific pathogen-free chickens in response to reticuloendotheliosis virus infection., 2017, 101(7): 2767-2777.
[6] Ojha C R, Rodriguez M, Dever S M, MUKHOPADHYAY R, EL-HAGE N. Mammalian microRNA: an important modulator of host-pathogen interactions in human viral infections., 2016, 23(1): 74.
[7] The Honeybee Genome Sequencing Consortium. Insights into social insects from the genome of the honeybee., 2006, 443(7114): 931-949.
[8] 刘芳. 意蜂哺育蜂与采集蜂头部mRNAs与miRNAs表达谱Solexa测序比较分析及其调控网络研究[D]. 杭州: 浙江大学, 2012.
LIU F. Integrating of Solexa high-abundance mRNAs and miRNAs in: comparison between nurses and foragers to identify regulatory network[D]. Hangzhou: Zhejiang University, 2012. (in Chinese)
[9] 郭昱, 苏松坤, 陈盛禄, 张少吾, 陈润生. LncRNA在蜜蜂级型分化中的功能研究. 生物化学与生物物理进展, 2015, 42(8): 750-757.
GUO Y, SU S K, CHEN S L, ZHANG S W, CHEN R S. The function of lncRNAs in the caste determination of the honeybee., 2015, 42(8): 750-757. (in Chinese)
[10] Ashby R, FORÊT S, Searle I, MALESZKA R. MicroRNAs in honey bee caste determination., 2016, 6: 18794.
[11] 陈晓. 蜜蜂卵巢激活和产卵过程差异表达的编码RNA与非编码RNA的筛选和鉴定[D]. 北京: 中国农业科学院, 2017.
CHEN X. Identification of differentially expressed coding and noncoding RNAs during ovary activation and oviposition in honey bees[D]. Beijing: Chinese Academy of Agricultural Sciences, 2017. (in Chinese)
[12] 石元元. 东方蜜蜂遗传图谱构建以及雌性蜜蜂发育分子机理[D]. 南昌: 江西农业大学, 2014.
SHI Y Y. Construction of genetic linkage map inand molecular mechanism of development in females honey bee[D]. Nanchang: Jiangxi Agricultural University, 2014. (in Chinese)
[13] 常伟. 蜜蜂消化道共生细菌及其多态性研究初探[D]. 福州: 福建农林大学, 2010.
CHANG W. Diversity of symbiotic bacteria in honeybee alimentary tract[D]. Fuzhou: Fujian Agriculture and Forestry University, 2010. (in Chinese)
[14] 贾慧茹. 亚致死剂量吡虫啉对意大利蜜蜂中肠菌群的影响[D]. 北京: 中国农业科学院, 2015.
JIA H R. Effect of the sublethal doses of imidacloprid on the bacterial diversity in the midgut of[D]. Beijing: Chinese Academy of Agricultural Sciences, 2015. (in Chinese)
[15] 郭睿, 解彦玲, 熊翠玲, 尹伟轩, 郑燕珍, 付中民, 陈大福. 意大利蜜蜂4、5和6日龄幼虫肠道发育过程中差异表达基因的趋势分析. 上海交通大学学报(农业科学版), 2018, 36(4): 14-21, 29.
GUO R, XIE Y L, XIONG C L, YI W X, ZHENG Y Z, FU Z M, CHEN D F. Trend analysis for differentially expressed genes in developmental process of 4-, 5- and 6-day-old larval guts of, 2018, 36(4): 14-21, 29. (in Chinese)
[16] Chen D, Guo R, Xu X, XIONG C l, LIANG Q, ZHENG Y z, LUO Q, ZHANG Z, HUANG Z J, KUMAR D, XI W J, ZOU X, LIU M. Uncovering the immune responses of, larval gut to, infection utilizing transcriptome sequencing., 2017, 621: 40-50.
[17] 陈大福, 郭睿, 熊翠玲, 梁勤, 郑燕珍, 徐细建, 黄枳腱, 张曌楠, 张璐, 李汶东, 童新宇, 席伟军. 胁迫意大利蜜蜂幼虫肠道的球囊菌的转录组分析. 昆虫学报, 2017, 60(4): 401-411.
CHEN D F, GUO R, XIONG C L, LIANG Q, ZHENG Y Z, XU X J, HUANG Z J, ZHANG Z N, ZHANG L, LI W D, TONG X Y, XI W J. Transcriptomic analysis ofstressing larval gut of(Hyemenoptera: Apidae)., 2017, 60(4): 401-411. (in Chinese)
[18] 王倩, 孙亮先, 肖培新, 刘锋, 康明江, 胥保华. 室内人工培育中华蜜蜂幼虫技术研究. 山东农业科学, 2009(11): 113-116.
WANG Q, SUN L X, XIAO P X, LIU F, KANG M J, XU B H. Study on technology for indoor artificial feeding oflarvae., 2009(11): 113-116. (in Chinese)
[19] Friedländer M R, Mackowiak S D, Li N, CHEN W, RAJEWSKY N. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades., 2012, 40(1): 37-52.
[20] Allen E, Xie Z, Gustafson A M, CARRINGTON J C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants., 2005, 121(2): 207-221.
[21] Smoot M E, Ono K, Ruscheinski J, WANG P L, IDEKER T. Cytoscape 2.8: new features for data integration and network visualization., 2011, 27(3): 431-432.
[22] Chen C, Ridzon D A, Broomer A J, ZHOU Z, LEE D H, NGUYEN J T, BARBISIN M, XU N L, MAHUVAKAR V R, ANDERSEN M R, LAO K Q, LIVAK K J, GUEGLER K J. Real-time quantification of microRNAs by stem-loop RT-PCR., 2005, 33(20): e179.
[23] Lucas K J, Zhao B, Liu S, RAIKHEL A S. Regulation of physiological processes by microRNAs in insects., 2015, 11: 1-7.
[24] Zhang Y, Zhou X, Ge X, LI M, JIA S, YANG X, KAN Y, MIAO X, ZHAO G, LI F, HUANG Y. Insect-specific microRNA involved in the development of the silkworm., 2009, 4(3): e4677.
[25] Liu S, Gao S, Zhang D, YIN J, XIANG Z, XIA Q. MicroRNAs show diverse and dynamic expression patterns in multiple tissues of., 2010, 11: 85.
[26] Liang P, Feng B, Zhou X G, GAO X W. Identification and developmental profiling of microRNAs in diamondback moth,(L.)., 2013, 8(11): e78787.
[27] 陈璇, 俞晓敏, 郑火青, 蔡亦梅, 胡福良. 西方蜜蜂(L.) sRNA的富集与文库检测. 中国农业科学, 2009, 42(8): 2943-2948.
CHEN X, YU X M, ZHENG H Q, CAI Y M, HU F L. Separation and enrichment of sRNAs from honeybee (L.) and its quality detection by library construction., 2009, 42(8): 2943-2948. (in Chinese)
[28] 陈璇. 蜜蜂() microRNA的全基因组挖掘及在雌性蜜蜂级型分化关键时期转录组水平调控作用[D]. 杭州: 浙江大学, 2012.
CHEN X. Genome-wide identification of microRNAs and their regulation of transcriptome on female caste determination of honey bee ()[D]. Hangzhou: Zhejiang University, 2012. (in Chinese)
[29] 汝玉涛, 王勇, 周敬林, 王德意, 马月月, 姜义仁, 高清, 秦利. 蜕皮激素受体和超气门蛋白基因在柞蚕发育过程及激素诱导后的表达模式. 蚕业科学, 2017(4): 594-602.
RU Y T, WANG Y, ZHOU J L, WANG D Y, MA Y Y, JIANG Y R, GAO Q, QIN L. The expression patterns of ecdysone receptor and ultraspiracle genes induring development and hormone-induced process., 2017(4): 594-602. (in Chinese)
[30] 舒旭. 家蚕蜕皮触发激素及其受体基因的克隆和表达分析[D]. 重庆: 西南大学, 2009.
SHU X. Cloning and expression analysis of genes encoding ecdysis triggering hormone and its receptor in the silkworm,[D]. Chongqing: Southwest University, 2009. (in Chinese)
[31] 周艳河. 白纹伊蚊中肠特异性高表达miRNA-miR-281对登革病毒复制的调节作用[D]. 广州: 南方医科大学, 2014.
ZHOU Y H. Dengue virus replication is regulated by miR-281: an abundant midgut-specific miRNA of vector mosquito[D]. Guangzhou: Southern Medical University, 2014. (in Chinese)
[32] 熊慧萍. 位于可变内含子区的果蝇microRNA-281-1/2基因转录和启动子分析[D]. 南京: 南京农业大学, 2008.
XIONG H P. Transcription and promoter analysis ofintronic microRNA-281-1/2 located in alternative spliced region[D]. Nanjing: Nanjing Agricultural University, 2008. (in Chinese)
[33] Ronshaugen M, Biemar F, Piel J, LEVINE M, LAI E C. ThemicroRNA iab-4 causes a dominant homeotic transformation of halteres to wings., 2005, 19(24): 2947-2952.
[34] TAMÁSI V, Monostory K, Prough R A, Falus A. Role of xenobiotic metabolism in cancer: involvement of transcriptional and miRNA regulation of P450s., 2011, 68(7): 1131-1146.
[35] 黄献彬. 小菜蛾解毒代谢相关miRNA鉴定及表达分析[D]. 福州: 福建农林大学, 2016.
HUANG X B. Identification and analysis of miRNAs involved in detoxification in the diamondback moth,[D]. Fuzhou: Fujian Agriculture and Forestry University, 2016. (in Chinese)
Differentially expressed microRNA and their regulation networks during the developmental process oflarval gut
Guo Rui1, Du Yu1, Xiong CuiLing1, Zheng YanZhen1, Fu ZhongMin1, Xu GuoJun1, Wang HaiPeng1, Chen HuaZhi1, Geng SiHai1, Zhou DingDing1, Shi CaiYun1, Zhao HongXia2, Chen DaFu1
(1College of Bee Science, Fujian Agriculture and Forestry University, Fuzhou 350002;2Guangdong Institute of Applied Biological Resources, Guangzhou 510260)
【Objective】MicroRNA (miRNA) is a kind of key regulator for negative regulation of mRNA at post-transcriptional level. The objective of this study is to provide miRNA expression patterns and differential expression information, illuminate the function of differentially expressed miRNA (DEmiRNA) in the development of larval gut by comprehensively investigating the DEmiRNAs and their regulation networks during the developmental process oflarval gut.【Method】Deep sequencing of the 4-, 5- and 6-day-old larval guts ofwas conducted using small RNA-seq (sRNA-seq) technology, followed by mapping of the data after quality-control with the reference genome of,and the mapped tags were then compared to miRBase database. The miRNA expression level was normalized by TPM algorithm, and the expression clustering, prediction of secondary structure of precursor and differential expression analysis were performed using related bioinformatic softwares. TargetFinder software was used to predict target gene of DEmiRNA, which was annotated to GO and KEGG databases using Blast, furthermore, miRNA-mRNA regulation networks were constructed using Cytoscape software. Stem-loop RT-qPCR was used to verify the sequencing data in this study.【Result】High-throughput sequencing of larval gut samples produced 10 841 644, 12 037 678 and 9 230 496 clean tags, respectively. In Am4 vs Am5 comparison group, there were16 up-regulated and 10 down-regulated miRNAs, while Am5 vs Am6 comparison groupincluded 5 up-regulated and 7 down-regulated miRNAs, respectively. Among them, novel-m0031-3p was shared by both Am4 vs Am5 and Am5 vs Am6, binding 5 target genes associated with ecdysone inducible protein, 25 and 11 DEmiRNAs were specific for the above-mentioned two comparison groups.DEmiRNA in Am4 vs Am5 could bind 5 742 target genes, among them 2 725 targets could be annotated to 46 GO terms in GO database, and the largest ones were binding, cellular process, metabolic process and single-organism process. similarly, 12 DEmiRNAs in Am5 vs Am6 could link 3 733 target genes, among them 2 725 targets could be annotated to 41 GO terms, and mostly enriched terms were binding, cellular process, single-organism processand metabolic process. In addition, 1 046 and 676 target genes of two comparison groups were related to 116 and 92 KEGG pathways, and the number of DEmiRNA target genes in Am4 vs Am5 was more than that in Am5 vs Am6, which annotated to Wnt signaling pathway, Hippo signaling pathway, purine metabolism and endocytosis. further analysis demonstrated that up-regulated and down-regulated miRNAs in Am4 vs Am5 could bind 611 and 85 target genes, and ame-miR-6052 linked the most target genes and participated in regulating cytochrome P450 via 5 target genes.miR-281-x could bind 49 target genes and indirectly regulate histidine metabolism, TGF-signaling pathway and Hippo signaling pathway.InAm5 vs Am6 comparison group, up-regulated and down-regulated miRNAs could bind 43 and 431 target genes, respectively, among them miR-iab-4-x linked the most target genes, and it could participate in regulating growth and development related pathways, such as dorso-ventral axis formation, Hippo signaling pathway, Wnt signaling pathway, FoxO signaling pathway, Notch signaling pathway and mTOR signaling pathway. Regulation network analysis indicated that complex networks formed between DEmiRNAs and target genes, and DEmiRNAs lied in the center while target genes lied in the periphery. Finally, Stem-loop RT-qPCR was carried out to validate the randomly selected three DEmiRNAs, and the result confirmed the reliability of sequencing data. 【Conclusion】The DEmiRNA and corresponding target genes in thelarval gut were predicted and analyzed at genome-wide level, it was found thatare capable of regulating the expression of many miRNAs such as ame-miR-6052, miR-iab-4-x, miR-281-x and novel-m0031-3p. The results not only offer the expression pattern and differential expression information of miRNA during the developmental process oflarval gut, but also lay a foundation for clarifying the molecular mechanisms underlying the larval gut’s development.
; larval gut; development; microRNA; target gene; regulation network
10.3864/j.issn.0578-1752.2018.21.018
2018-05-22;
2018-06-28
国家自然科学基金(31702190)、国家现代农业产业技术体系建设专项资金(CARS-44-KXJ7)、福建省科技计划项目(2018J05042)、福建省教育厅中青年教师教育科研项目(JAT170158)、福建农林大学科技创新专项基金(CXZX2017343)、福建农林大学科技发展基金(KF2015123)
郭睿,E-mail:ruiguo@fafu.edu.cn。杜宇,E-mail:m18505700830@163.com。郭睿和杜宇为同等贡献作者。通信作者陈大福,E-mail: dfchen826@fafu.edu.cn
(责任编辑 岳梅)
猜你喜欢
蜜蜂杂志(2021年6期)2021-12-05
信阳农林学院学报(2021年1期)2021-04-01
林业科技(2020年3期)2021-01-21
蜜蜂杂志(2020年6期)2020-12-02
养猪(2020年1期)2020-02-19
蜜蜂杂志(2019年3期)2019-12-30
今日农业(2019年11期)2019-08-15
中国蜂业(2019年8期)2019-01-08
国外畜牧学·猪与禽(2018年11期)2018-05-14
农村农业农民·B版(2016年7期)2016-10-21
