
一种合成超顺磁CaFe2O4纳米颗粒的新方法及其络合机理*
2018-11-15 11:05:52赵红建
无机盐工业 2018年11期
马 富,赵红建
(宁夏师范学院化学化工学院,宁夏固原756000)
MFe2O4(M=Mn2+、Ni2+、Fe2+、Co2+、Mg2+、Ca2+、Ba2+)铁酸盐是一系列重要的多功能磁性材料,由于它们特殊的物理化学性质,在生物医药、新的给药系统材料、药物输送、微波器件、高密度磁存储系统以及催化剂等领域具有广阔的应用前景,受到了科研人员的广泛关注[1-6]。其中,CaFe2O4是一种尖晶石型铁酸盐磁性材料,已在微电子领域应用超过半个世纪[7]。通常生物医药、给药系统材料以及药物输送均要求所输送的粒子表现为顺磁性行为,然而正交的CaFe2O4块材在室温表现出铁磁行为,因此需要开发一种高效的制备方法来合成纳米尺寸的CaFe2O4顺磁性材料[8]。此外,纳米磁性材料在制备过程中,由于磁性粒子间的相互作用颗粒容易产生团聚,导致所制备的磁性材料的磁学性能降低,为合成纳米磁性材料带来了巨大的挑战。因此,开发一种制备高分散CaFe2O4磁性材料的方法并研究它的磁学性质具有重要的意义。
近年来,多种方法被用来制备尖晶石型CaFe2O4铁酸盐磁性材料,包括微波烧结技术[9]、溶胶-凝胶法[10]、聚合物前驱体法[11]、溶液法[3,12]以及聚丙烯酰胺凝胶法[13-14]等方法。在这些方法中,聚丙烯酰胺凝胶法是一种非常有效的方法,可通过改变螯合剂、有机添加剂、pH、丙烯酰胺与交联剂的比例、聚合方式以及添加容易去除的添加剂等手段获得高分散的磁性纳米颗粒[8]。 自 1989 年 A.Douy 等[15]首次采用这一方法制备YBa2Cu3O7-x超导体的超细粉末以来,该方法得到了突飞猛进的发展。随着科学技术的进步与发展,该方法也在不断改进,已由温度控制聚合反应代替添加聚合引发剂发生聚合反应来合成金属氧化物纳米颗粒[8]。研究发现,不同阴离子种类对制备的ZnAl2O4纳米颗粒的相纯度、形貌以及物理化学性质有极大的影响[16]。目前,聚丙烯酰胺凝胶法已被广泛用于制备陶瓷粉末、发光材料、多铁性材料、热电材料、固体氧化物电池材料、光催化材料以及纳米复合材料等[8]。
但是,在通过聚丙烯酰胺凝胶法制备磁性材料上进展并不顺利,研究人员尝试对该方法加以改进,以期获得高分散的磁性纳米颗粒。赵东方等[17]以乙二胺四乙酸二钠(EDTA)和乙酸作为络合剂制备不同的NiFe2O4时,获得的纳米颗粒团聚现象比较严重。W.P.Wang等[18]以EDTA作为络合剂,通过改变丙烯酰胺与亚甲基双丙烯酰胺的比例来制备CoFe2O4,获得的纳米颗粒细小,并出现了少量的团聚。S.F.Wang等[13]采用聚丙烯酰胺凝胶法制备了(Mg,Ca,Ba)Fe2O4磁性纳米颗粒,获得的纳米颗粒团聚现象较明显,且其磁学性质受到一定程度的影响。本研究采用超声辅助聚丙烯酰胺凝胶法的方法制备CaFe2O4纳米颗粒,并对其络合机理、相纯度、形貌以及磁学性质做了研究。
1 实验
1.1 样品制备
按化学计量比称取一定量的碳颗粒、硝酸钙和硝酸铁,在超声搅拌下,先将碳颗粒用酒精分散,清洗数次,再用去离子水清洗数次;缓慢加入1.6 mol/L的稀硝酸溶液,再逐次加入硝酸钙和硝酸铁,待二者完全溶解,加入与钙离子与铁离子物质的量比为1.5∶1的柠檬酸作为络合剂。待柠檬酸完全溶解后,加入20 g葡萄糖,使其在凝胶干燥过程中不至于导致凝胶塌缩。随后,按与金属阳离子的物质的量比为9∶1的比例,加入丙烯酰胺单体,每步均在超声搅拌中进行,以使烧杯中的添加物充分溶解。待上述溶液混合均匀后,用氨水将整个溶液的pH调为3。最后,将所得褐色溶液加热至到95℃,使丙烯酰胺与亚甲基双丙烯酰胺发生热聚合反应,20 min后,溶液缓慢转变为褐色凝胶体。将获得的褐色凝胶放在恒温干燥箱中,在135℃恒温干燥24 h,最终形成黑色干凝胶。将黑色干凝胶研成细粉,置入箱式炉中在600℃烧结5 h,得到CaFe2O4纳米颗粒。
1.2 样品测试与表征
利用DX-2700型X射线衍射仪对CaFe2O4纳米颗粒的物相进行分析;利用ULTRA 55型场发射扫描电子显微镜观察CaFe2O4纳米颗粒的表面形貌;利用STA 449C型同步热分析仪的差热分析法对CaFe2O4干凝胶的热降解过程进行分析;利用SQUID型超导量子干涉仪磁性测量系统测量样品的磁性。
2 结果与讨论
2.1 络合机理分析
聚丙烯酰胺凝胶法在合成金属氧化物纳米颗粒的过程中,一般会经历3个反应[19]:一个是金属离子与络合剂之间的反应,称为络合反应;一个是丙烯酰胺与亚甲基双丙烯酰胺之间的反应,称为交联反应;另一个是丙烯酰胺与交联反应的产物之间的聚合反应。其中,交联反应和聚合反应并不会改变金属阳离子最终的配位情况,即如无中间产物的影响不会导致最终产物的相结构发生变化,其主要影响最终产物的形貌[20]。因此,对聚丙烯酰胺凝胶法的机理分析主要集中在络合反应。图1是柠檬酸络合钙离子、铁离子的络合反应示意图。图1a是柠檬酸的分子结构式,它是一羟基和羧基复合型的络合剂,单个的柠檬酸分子由3个羧基和1个羟基组成。当柠檬酸与金属离子发生络合反应时,金属离子首先与羧基反应,只有当溶液中的金属离子还需要羟基进行反应时羟基才参与反应。在尖晶石型CaFe2O4中,Ca2+的配位数是8,而Fe3+的配位数是6,在发生络合反应时,Ca2+和Fe3+分别会与柠檬酸中的羧基或者羟基络合,最终形成单个分子单元。图1b是柠檬酸与钙离子、铁离子发生络合反应时的结构示意图,从图1b可以看出,5个柠檬酸分子以羧基和羟基中的—OH与金属离子配位,形成了单个的分子单元。最终与碳颗粒一起,被限制在丙烯酰胺与亚甲基双丙烯酰胺聚合形成的三维网状结构中。当在高温烧结去除有机物时,碳颗粒也被去除,最终形成了高分散的CaFe2O4纳米颗粒。

图1 柠檬酸的结构式(a)和柠檬酸络合钙离子、铁离子的结构式(b)
2.2 相结构分析
为了研究CaFe2O4纳米颗粒的成相温度,对CaFe2O4干凝胶做了差热(DTG)分析,结果见图2。从图2可以看出,整个过程包括5个放热反应。第一个放热反应峰位于211℃附近,主要归因于干凝胶中结构水的蒸发或分解[21-22];第二个放热峰位于303℃附近,主要归因于小分子有机基团的分解[23-24];第三个放热峰位于403℃附近,主要归因于络合的COO—基团和残余的C=O基团等的分解[25];第四个放热峰位于469℃附近,主要归因于高分子网络凝胶体的侧链的分解[20-22];第五个放热过程最为显著,放热峰峰值位于504℃附近,主要归因于碳质骨架、主链以及其他有机残留物的氧化分解和结晶放热过程[23-26]。当温度高达520℃时,再无明显的放热峰出现,表明前驱体中的有机物已被完全去除。
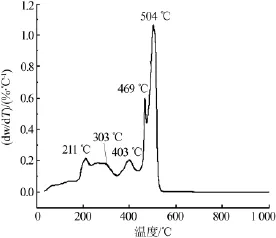
图2 CaFe2O4干凝胶的DTG曲线
图3是CaFe2O4干凝胶在600℃烧结所得产物的XRD谱图。从图3可见,在衍射峰25.497、30.203、31.756、33.471、35.466、39.044、40.301、42.561、45.123、46.358、49.583、51.979、52.347、55.150、59.878、61.427、63.838°处对应的晶面指数分别为(220)、(011)、(111)、(040)、(121)、 (031)、 (131)、(420)、 (321)、(141)、(241)、(510)、(051)、(260)、(360)、(170)和(261),对照 JCPDS 标准卡片(32-0168)属于正交CaFe2O4相。由XRD结果可知,采用超声辅助聚丙烯酰胺凝胶法制备CaFe2O4纳米颗粒的成相温度比采用聚丙烯酰胺凝胶法要低200℃[13]。根据文献[16],相关的反应式:
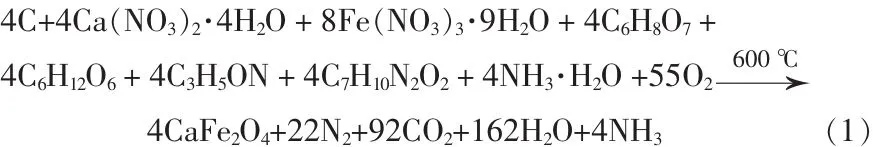

图3 CaFe2O4干凝胶在600℃烧结所得产物的XRD谱图
2.3 XPS分析

图4 CaFe2O4干凝胶在600℃烧结所得产物的XPS谱图
为了进一步分析CaFe2O4干凝胶在600℃烧结所得产物的相纯度,图4为产物的XPS谱图。由图4a的CaFe2O4纳米颗粒XPS全谱图可以看出,除了O、C、Ca和Fe元素外,产物不含其他元素,表明在合成CaFe2O4纳米颗粒的整个过程中,没有引入别的杂质元素(如N等)。从图4b可以看出,Ca2p能级被分离为 Ca 2p3/2(343.45 eV)和 Ca 2p1/2(347.59 eV)两个组分,可归因于正交CaFe2O4中的Ca2+。在图4c中,Fe 2p 能级也能分离成 Fe 2p3/2(707.21 eV)和Fe2p1/2(721.31eV)两个组分,可归因于正交 CaFe2O4中的Fe3+。通常情况下,在O 1s谱图中如果出现金属—OH的峰,则表明所制备的目标产物是亲水性的,进而可以判断样品中可能会含有少量的氢氧化物。在本实验中,仅有一个明显的特征峰(约527.64 eV)出现在样品中(图4d),这可能是由Fe—O或Ca—O键所引起的。基于XRD和XPS实验结果可知,干凝胶在600℃烧结所得产物属于纯相的正交尖晶石型CaFe2O4。
2.4 形貌分析
图5是CaFe2O4干凝胶在600℃烧结所得产物的SEM照片。由图5可以看出,颗粒呈近似球形,且细小均匀,颗粒间无黏连团聚现象。
图6为CaFe2O4纳米颗粒的尺寸分布曲线。由图6可见,产品的平均颗粒尺寸约为30 nm。与传统的聚丙烯酰胺凝胶法相比,由于在前驱体溶液中引入了容易去除的碳颗粒,在丙烯酰胺与亚甲基双丙烯酰胺聚合形成三维网络结构的过程中,碳颗粒和金属络合物一起被限制在三维网络结构内,很大程度上阻止了粒子之间在溶液中的大量移动和聚集,因此更有利于合成出高分散的CaFe2O4纳米颗粒[13]。此外,碳颗粒的引入,更容易导致所制备产物颗粒的细化[27]。

图5 CaFe2O4干凝胶在600℃烧结所得产物的SEM照片

图6 CaFe2O4纳米颗粒的 颗粒尺寸分布曲线
2.5 磁性分析
将CaFe2O4纳米颗粒在零场下冷却到-263.15℃,外加50Oe的磁场,并以1℃/min将温度升至26.85℃,测量其直流磁化强度随温度的变化趋势,称为零场冷却曲线(ZFC);随后,保持外加磁场不变,将CaFe2O4纳米颗粒冷却至-263.15℃,随温度升高并再次测量其磁化强度随温度的变化曲线,即场冷却曲线(FC),结果如图 7a所示。从图 7a可见,CaFe2O4纳米颗粒在低温存在一个阻挡温度 (TB=-173.15℃),该数值比文献[28]报道的略小,这一阻挡温度可能是由于纳米颗粒之间的热能和磁晶各向异性能相互竞争所致。从图7a还可以看出,ZFC曲线出现了明显的峰,可进一步区分样品的顺磁态与温度之间的函数关系。
为了进一步讨论CaFe2O4体系中磁相变随温度的变化关系,图7b给出了CaFe2O4纳米颗粒直流磁化率倒数1/χ随温度的变化关系曲线。由图7b可知,在低温区(T<-228.15℃)服从居里-外斯定理:

式中,χ是磁化率,emu/g;T 是温度,℃;θCW是居里-外斯温度,℃;C是居里常数。根据拟合结果,CaFe2O4纳米颗粒的θCW大约为-335.15℃,负值表明样品的反铁磁相互作用占支配地位。

图7 CaFe2O4干凝胶在600℃烧结所得产物的场冷却(FC)和零场冷(ZFC)却曲线
图8是CaFe2O4纳米颗粒在外加磁场50 Oe下不同温度的磁滞回线。由图8a可知,在-272.15℃时,磁滞回线表现出明显的回线特征,表明CaFe2O4纳米颗粒在低温下呈现弱的铁磁性;在26.85℃时,样品呈现顺磁行为(图8b),其结果与图7所得的分析结果相吻合。根据文献[28]可知,块体的CaFe2O4在室温表现出弱的铁磁行为,而纳米尺寸的样品表现出顺磁性,这一结果与本实验的结果一致。

图8 CaFe2O4干凝胶在600℃烧结所得产物在不同温度下的磁滞回线
3 结论
采用超声辅助聚丙烯酰胺凝胶法合成了CaFe2O4纳米颗粒, 利用 DTG、XRD、XPS、SEM 以及SQUID等手段对样品做了表征。结果表明,干凝胶在600℃烧结获得了纯相的CaFe2O4纳米颗粒。颗粒粒度细小,均匀分散,平均粒径约为30 nm。磁性分析发现,其在-173.15℃左右存在一个阻挡温度。在阻挡温度以上,CaFe2O4纳米颗粒表现出顺磁行为,在药物输运领域具有潜在的应用。基于实验结果,分析了柠檬酸络合金属离子的络合机理。
猜你喜欢
云南化工(2021年11期)2022-01-12 06:06:18
江苏调味副食品(2021年1期)2021-04-01 12:51:12
长春理工大学学报(自然科学版)(2019年4期)2019-09-02 09:18:06
新型建筑材料(2018年5期)2018-06-14 06:15:02
现代食品(2016年24期)2016-04-28 08:12:08
当代化工研究(2016年2期)2016-03-20 16:21:20
科技资讯(2015年21期)2015-11-14 19:46:21
储能科学与技术(2015年4期)2015-11-14 00:48:24
应用化工(2015年2期)2015-07-13 03:12:26
应用化工(2014年1期)2014-08-16 13:34:08
