
复合稀释剂体系下聚偏氟乙烯平板膜的制备与表征
2018-11-10 03:50史梦芝张宇峰
天津工业大学学报 2018年5期
王 薇 ,史梦芝 ,张宇峰
(1.天津工业大学 省部共建分离膜与膜过程国家重点实验室,天津 300387;2.天津工业大学 材料科学与工程学院,天津 300387)
聚偏氟乙烯(PVDF)具有优良的力学性能、抗冲击性、耐磨性以及稳定的化学性能,被视为一种制备分离膜的良性材料.目前用于制备PVDF膜的方法有多种,常用的方法有非溶剂致相分离(NIPS)法和热致相分离(TIPS)法[1-2].吕晓龙等[3]介绍了利用NIPS法制备PVDF超滤膜的相分离原理、膜结构控制方法及膜应用等关键研究进展;欧洋等[4]以聚丙烯腈/二甲基亚砜/N,N-二甲基甲酰胺三元体系制备得到聚丙烯腈多孔超细纤维,并提出非溶剂致相分离是主要的成孔机理;王薇[5]等采用NIPS法,添加羧基化多壁碳纳米管(MWCNTs-COOH)制备PVDF中空纤维超滤膜,研究了MWCNTs-COOH的添加量、管径对超滤膜性能的影响.但是NIPS法需要大量使用溶剂,必须对溶剂进行回收和再利用,而且制得的膜力学性能较差[6-8].
TIPS法是由温度作为相分离的驱动力,将体系升温至聚合物熔点以上,使聚合物与稀释剂混合形成均相铸膜液,再对体系进行降温冷却,铸膜液分相,固化成膜[9].冯栓虎[10]使用离子液体替代有机溶剂作为稀释剂,采用TIPS法制备了PVDF微孔膜,并研究了成膜机理;王世乾[11]以邻苯二甲酸二丁酯(DBP)与已二酸二辛脂(DOA)组成混合稀释剂,通过TIPS法制备了PVDF平板膜并研究了稀释剂配比、固含量变化以及无机粒子对膜微观结构与性能的影响.相对于NIPS法而言,TIPS法中的工艺参数较少,使得制膜过程更好控制且稳定.但是,用TIPS法制膜时,体系温度较高,能耗较大.为了减少不必要的能耗,本文采用低温热致相分离(L-TIPS)法制膜,即将铸膜液温度控制在铸膜液浊点温度与聚合物熔点温度之间,同时要求凝固浴温度显著低于铸膜液的浊点温度.这样,当铸膜液进入凝固浴时,就会同时发生热致相分离和非溶剂致相分离[12-13].
邻苯二甲酸二丁酯(DBP)作为PVDF的良溶剂[14-20],与PVDF之间的相互作用较强,体系易发生固-液相分离,容易形成松散的球粒堆积结构,使膜的力学性能降低.由于邻苯二甲酸二辛酯(DOP)与DBP的性质相似,且与PVDF相互作用较弱,与PVDF/DBP体系混合时不会发生其他副反应,而且能够改变PVDF与稀释剂之间的相溶性,从而改变体系的相分离方式,得到性能较优的分离膜.因此选择DBP/DOP作为复合稀释剂.
本文选择PVDF作为基体材料,研究PVDF的浓度、复合稀释剂DBP/DOP的配比、降温速率以及凝固浴温度对膜结构与性能的影响.
1 实验部分
1.1 实验试剂与仪器
聚偏氟乙烯(PVDF),Solvay公司产品,使用前需在120℃下烘干;邻苯二甲酸二丁酯(DBP)、邻苯二甲酸二辛酯(DOP)、无水氯化锂(LiCl)、无水乙醇(C2H6O),均为工业纯,天津市科密欧化学试剂有限公司生产;次氯酸钠(NaClO)、聚乙二醇(PEG400、PEG10000),均为分析纯,天津市科密欧化学试剂有限公司生产;甘油,化学纯,天津市化学试剂三厂生产;牛血清蛋白(BSA),分析纯,北京市普博欣生物科技有限公司生产;去离子水,实验室自制.
JA3003型电子天平,天津市天马仪器厂产品;电热套,河南爱博特科技有限发展公司产品;DW-11型无级调速增力搅拌器:巩义市予华仪器有限公司产品;玻璃板,实验室自备;刮膜棒,市售;分离膜评价装置,实验室自组装;JBDL-200N型电子拉力试验机,市售;UV754PC型紫外分光光度计,上海佑科仪器有限公司产品;浊点测试仪,实验室自制;DSA100型接触角测量仪,德国KRUSS公司产品;Hitachi S-4800型场发射扫描电镜,日本日立公司产品;外径千分尺,上海衡器量器厂产品;SNB-1型数字粘度计,上海精科天美科学仪器有限公司产品.
1.2 PVDF平板膜的制备
分别准确称取质量分数为20%、25%、30%、35%、40%的PVDF树脂,将其与LiCl、PEG400和复合稀释剂(配比见表1)置于干燥的三口烧瓶中,在一定的温度下机械搅拌5 h,待PVDF溶解后得到稳定、均一的铸膜液.铸膜液形成后,停止搅拌,静置,保温脱泡4 h.而后将适量铸膜液流延到洁净、干燥的玻璃板上,用刮膜棒将铸膜液刮出具有一定厚度的薄层;然后迅速将玻璃板放到去离子水中,待膜冷却固化,从玻璃板上自然脱落;将制备好的平板膜置于去离子水中24 h,以去除膜中残留的稀释剂和添加剂;再将其转移至甘油溶液中浸泡24 h,以防止膜孔的塌陷,保存待测试.
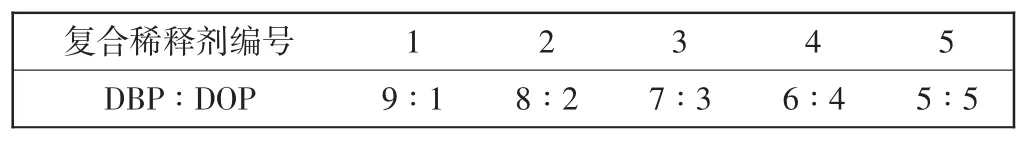
表1 DBP/DOP质量比Tab.1 DBP/DOP mass ratio
1.3 PVDF平板膜的结构表征与性能测试
(1)形貌结构.将制备好的平板膜置于105℃真空烘箱中干燥12 h,后用液氮将其淬断,经真空喷金处理后,置于场发射扫描电子显微镜下进行观察和拍照,调节放大倍率观察膜的断面形貌及膜的表面形貌.
(2)浊点温度.取少量混合物试样置于加热台上缓慢升温至200℃,使试样保持熔融状态并保温一段时间,待试样完全透明后,以一定速率降温,待试样中出现浑浊时记录此时的温度,即为浊点温度.
(3)纯水通量.采用实验室自制的分离膜性能测试装置测试平板膜的水通量.首先用去离子水将膜片润湿,在0.1 MPa的压力下预压20 min至水通量基本稳定,然后在恒压状态下,用量筒接取一定量的透过液,记录透过液的体积和测试时间,并测量膜片的有效表面积,测试3次求取平均值.纯水通量的计算公式如下:

式中:F为膜的纯水通量(L/(m2·h));V为透过液的体积(L);A 为膜片的有效表面积(m2);t为测试时间(h).
(4)截留率.采用实验室自制的分离膜性能测试装置测试平板膜的截留率.将PEG10000配成质量浓度为5 000 mg/L的原液,并在0.1 MPa的压力下通过平板膜组件,预压20 min至水通量基本稳定,测量原液和透过液在波长为510 nm处的吸光度值,通过标准曲线得到原液和透过液的质量浓度.截留率计算公式如下:

式中:R为膜片的截留率(%);Cp为透过液质量浓度(g/L);C0为原液质量浓度(g/L).
(5)力学性能.将平板膜剪成合适尺寸,然后通过JBDL-200N型电子拉力试验机测试所制备样品的拉伸强度(MPa)和断裂伸长率(%).夹距 100 mm,拉伸速率5 mm/min,每个样品测试5次,取平均值.
(6)孔隙率及平均孔径.取适量膜片,用去离子水充分浸润后,用滤纸将膜片表面的水分吸干,用电子天平称出膜的湿态质量,在105℃的真空干燥箱中干燥24 h,准确称量膜的干态质量,从而计算出膜的孔隙率ε.计算公式如下:
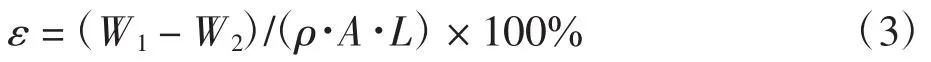
式中:ε为平板膜的孔隙率(%);W1为膜片的湿态质量(g);W2为膜片的干态质量(g);ρ为 PVDF 的密度(g/cm3);A 为膜片的有效表面积(cm2);L 为膜片的厚度(cm).利用上述数据,代入公式(4)即可计算出平板膜的平均孔径:
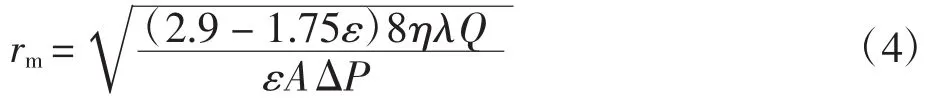
式中:rm为平板膜的平均孔径(m);η为透过液的黏度(Pa·s);λ 为膜厚(m);Q 为水通量(m3/s);A 为膜的有效表面积(m2);ΔP 为加在膜两侧的压力(Pa).
2 结果与讨论
2.1 PVDF的浓度对体系及成膜的影响
2.1.1 PVDF的浓度对体系粘度的影响
体系粘度反映的是高分子链段间相互运动时剪切力的大小.凝固浴中溶剂交换速率的大小受体系粘度的影响,通过改变溶剂的交换速率进而改变分离膜的结构与性能.本文测试了体系粘度与PVDF浓度的变化关系,如图1所示.
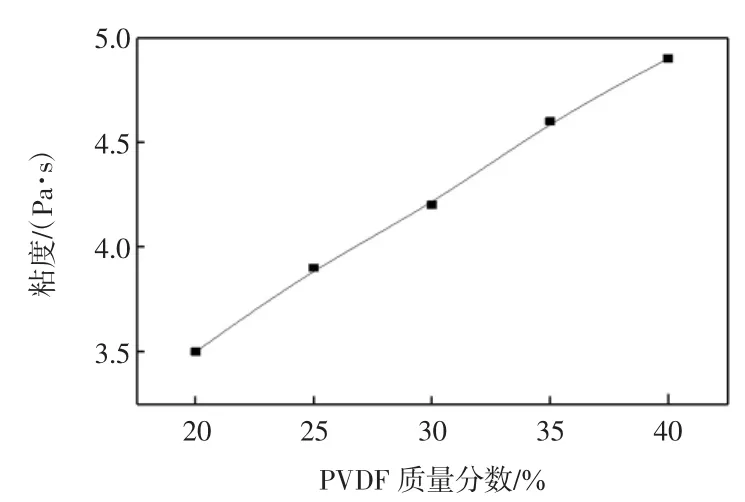
图1 体系粘度随PVDF质量分数的变化关系Fig.1 Variation of viscosity with different PVDF mass fractions
由图1可知,体系的粘度随PVDF质量分数的增大而有所提高.这是由于聚合物浓度的增加使得聚合物分子数量增加,分子间的相互作用力也随之增大,分子在做无规则运动时会产生更高的内摩擦力,从而使得体系的粘度增加.
2.1.2 PVDF的浓度对膜微观结构的影响
其他实验条件相同而仅改变体系中PVDF浓度时,得到的膜断面及局部放大的结构如图2所示.
由图2可以看到,指状大孔结构与海绵状结构共同存在,说明在相分离过程中既包含热致相分离,又包含非溶剂致相分离.图2(a)中,膜在高倍放大后,球粒结构表面呈绒毛状,且相邻结构之间的间隙较大,生成的球晶不规整;图2(b)中,球晶表面变得光滑,球粒之间的孔隙相对较小,球粒堆积相对较密,球晶生长规整;图2(c)中,球晶堆积非常紧密,球晶之间的边界变得模糊,球粒相互接触,有向连续体发展的趋势.由图2(a)—图2(c)可以看出,膜的球粒结构从松散的绒毛状结构先过渡为较密的球粒堆积结构,再逐渐向双连续结构演变.这是由于聚合物浓度的升高使得聚合物分子的数量在一定程度上增加,体系的粘度随之增大,造成在冷却过程中,膜内的晶核密度增大,最终使得球晶的数量增加,尺寸减小.

图2 不同PVDF浓度下膜的电镜图Fig.2 SEM images of membranes containing different PVDF concentrations
2.1.3 PVDF的浓度对膜纯水通量和截留率的影响
考察平板膜的纯水通量、对PEG10000的截留率以及膜的孔隙率随PVDF浓度的变化关系,结果分别如图3和图4所示.
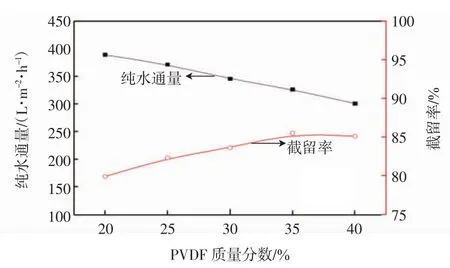
图3 纯水通量和PEG10000截留率随PVDF浓度的变化关系Fig.3 Pure water flux and PEG10000 rejection rate with different PVDF concentrations
由图3和图4可知,膜的纯水通量和孔隙率均随着PVDF质量分数的增大而减小,截留率则随着PVDF质量分数的增大而增大.这是因为体系中聚合物浓度的增加使得在冷却过程中膜内的晶核密度增大,晶核排列变得更加紧密,使得孔隙率在一定程度上降低,纯水通量也随之降低;另一方面由于聚合物浓度的增加会堵塞一部分膜孔,使膜孔由最初的开放状态转变为闭合状态,孔之间的连通性变差,导致平板膜孔隙率下降,水通量降低.
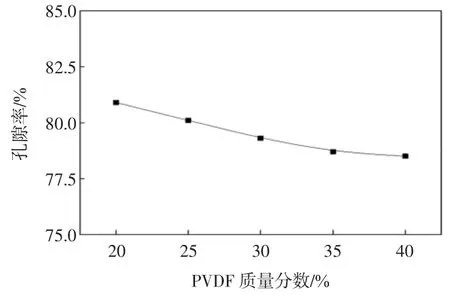
图4 膜孔隙率随PVDF浓度的变化关系Fig.4 Membrane porosity with different PVDF concentrations
2.1.4 PVDF的浓度对膜力学性能的影响
平板膜的力学性能随PVDF浓度的变化关系如图5所示.
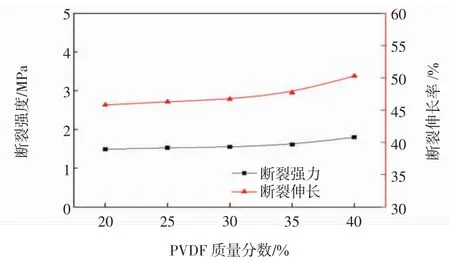
图5 不同浓度PVDF膜的力学性能Fig.5 Mechanical properties of membranes with different PVDF concentrations
由图5可知,膜的拉伸强度和断裂伸长率均随着PVDF浓度的增加而增加.由图2电镜图片可以看出,PVDF为低浓度时制备得到的平板膜的膜结构相对松散,球粒之间的间隙较大,相应的孔隙率也较高,故拉伸强度和断裂伸长率均低;而用高浓度的PVDF制备的平板膜,由于膜结构变得致密,膜孔隙率大大降低,使得膜的拉伸强度和断裂伸长率均存在不同程度的上升.这一结果表明聚合物的浓度影响膜的力学性能.
2.2 稀释剂配比对体系及成膜的影响
2.2.1 稀释剂配比对体系浊点温度的影响
图6所示为稀释剂配比对体系浊点温度的影响.
由图6可以看出:当DBP∶DOP质量比为9∶1时,体系的结晶温度会随着PVDF浓度的升高而升高,几乎呈线性关系,表明铸膜液在冷却降温、固化成膜的过程中遵循固-液相分离机理.含量较少的弱稀释剂对PVDF与强稀释剂之间的相互作用影响很小.然而,随着弱稀释剂含量的逐渐增大,当PVDF处于低浓度时,其结晶温度相对稳定,一旦体系温度高于PVDF的结晶温度时,便会出现浊点,并且体系的浊点温度和PVDF的结晶温度均会随着弱稀释剂含量的增加而有所上升,只是相比于浊点温度,PVDF的结晶温度上升幅度较小.弱稀释剂含量的增加,增大了PVDF与复合稀释剂之间的相互作用参数,使得PVDF在稀释剂中的相溶性大大减弱,此时体系会由单一的固-液相分离转变为固-液相分离和液-液相分离并存的情形,而且液-液相分离区也会增大.综上所述,可以通过改变稀释剂的组成及配比,调控聚合物与稀释剂之间的相互作用参数来达到控制相分离过程的目的,使之朝着有利于制备高性能分离膜的方向发展.
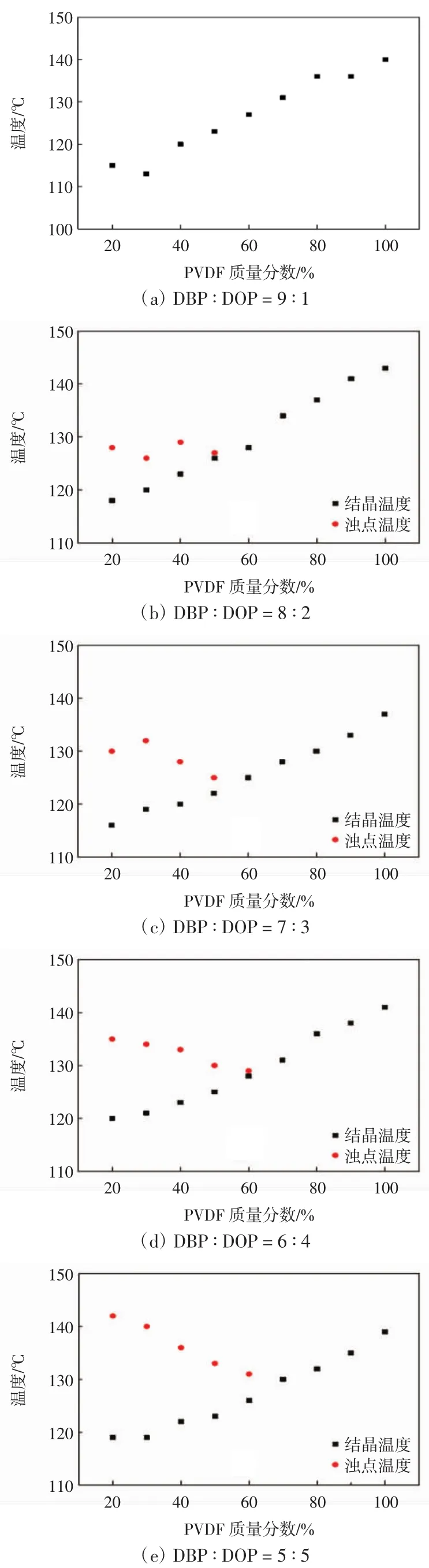
图6 PVDF/DBP/DOP体系热力学相图Fig.6 Thermodynamic phase diagram of PVDF/DBP/DOP system
2.2.2 稀释剂配比对体系粘度的影响
PVDF质量分数为25%时不同稀释剂配比下体系粘度的变化情况如图7所示.
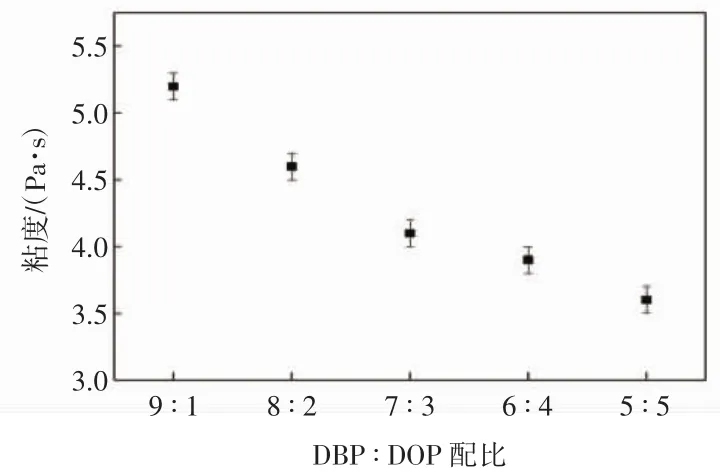
图7 体系粘度随不同稀释剂配比的变化关系图Fig.7 Variation of viscosity with different concentration ratio of diluent
由图7可以看出,当PVDF的浓度一定且铸膜液温度恒定时,PVDF/DBP/DOP体系的粘度随着DOP含量的增加而降低.这是因为DOP的加入,降低了PVDF与复合稀释剂之间的相溶性,相互作用参数增大,造成体系的粘度降低.
2.2.3 稀释剂配比对膜微观结构的影响
图8所示为不同稀释剂配比下PVDF/DBP/DOP体系经20℃水浴冷却后得到的膜局部结构放大图.
由图8可知,在制膜条件及工艺相同的情况下,DBP∶DOP为9∶1时,球粒堆积结构明显,且球粒表面具有绒毛状孔结构,球粒之间边界清晰;随着DOP含量的增加,虽然体系中仍存在球晶结构,但痕迹越来越模糊,球晶的边界和球粒之间的孔隙逐渐消失,成为连续体;随着DOP含量的进一步增大,球晶结构完全消失,体系中出现了双连续结构.从图6热力学相图可以看出,体系中DBP∶DOP质量比为9∶1时,冷却降温过程中的相分离遵循固-液分相机理,会有大量的PVDF分子结晶,随着结晶的增多,其生长前沿的铸膜液浓度会大幅降低,从而出现了具有绒毛状形态的球粒结构;当DBP∶DOP质量比为7∶3时,PVDF与复合稀释剂之间的相溶性降低,表现为体系浊点温度的升高.在冷却降温过程中,体系首先经历液-液分相,球晶的生长受到极大的抑制,但由于凝固浴温度显著低于PVDF的结晶温度,故经历液-液相分离后随之而来的便是PVDF的结晶过程,所以在膜中仍能看到模糊的球晶结构;而当DBP∶DOP质量比为5∶5时,体系的浊点温度升高幅度较大,液-液相分离区域也随之增大,液-液分相向固-液分相的转变时间延长,PVDF的结晶过程受阻,膜中出现了双连续结构.

图8 不同稀释剂配比下的膜结构Fig.8 Structures of membranes with different concentration ratio of diluent
2.3 降温速率对膜结构及性能的影响
2.3.1 降温速率对膜微观结构的影响
采用两种不同的冷却方式(20℃水浴冷却和20℃空气冷却,其中水冷速度大于气冷)来研究不同的降温速率对膜微观结构的影响,结果如图9所示.

图9 不同降温速率下的膜结构Fig.9 Structures of membranes under different cooling rate conditions
由图 9可见,图 9(a)和图 9(b)中均为球晶结构,但图9(a)中球晶的规整度较差,而且尺寸较小.这是由于两个体系均是先经历液-液分相,再经历固-液分相,但在图9(a)中,由于水浴冷却速率较快,使得完成两个相分离阶段的用时很短,在球晶还没生长完全时,相分离过程已经提前结束;图9(b)则恰恰相反,空气冷却速率较慢,为球晶生长提供了充足的时间,球晶形态可以逐渐完善,球晶尺寸也相对较大.
2.3.2 降温速率对膜性能的影响
表2所示为相同制膜条件及工艺下,仅改变降温速率对膜的水通量、截留率、孔隙率和平均孔径的影响,其中,PVDF的浓度及复合稀释剂的比例固定.
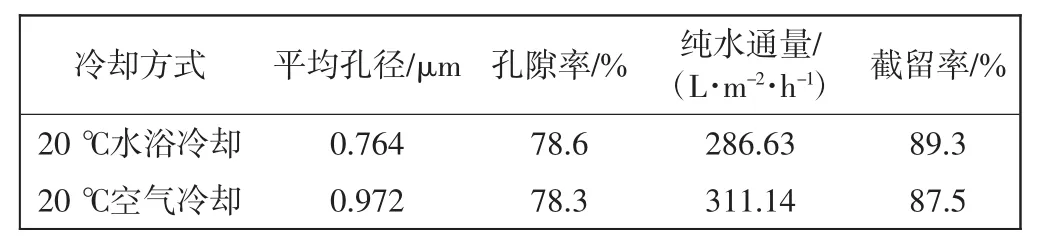
表2 降温速率对膜性能的影响Tab.2 Effect of cooling rate on performance of membranes
由表2可知,在20℃空气冷却下膜纯水通量较高,截留率较低,而在两种冷却方式下,膜的孔隙率变化却不太大.可以解释为缓慢的降温速率会延长PVDF的结晶过程及晶体的粗化过程所需的时间,相分离过程进行得彻底,得到的晶体尺寸和膜孔孔径较大,但是孔隙率的大小受聚合物浓度的影响,在聚合物浓度一定的情况下,膜的孔隙率几乎没有变化.由此可知,降温速率不会影响膜的孔隙率.在孔隙率不变,平均孔径变大的情况下,降温速率越慢,膜的纯水通量越大,相应的截留率会有所降低.
2.4 凝固浴温度对膜结构及性能的影响
2.4.1 凝固浴温度对膜微观结构的影响
凝固浴温度对膜结构的影响如图10所示.

图10 凝固浴温度对膜结构的影响Fig.10 Effect of coagulation bath temperature on structures of membranes
由图10可以看出,图10(a)为不规则的球粒堆积结构,图10(b)为双连续结构,且具有球晶结构的痕迹.这是由于在20℃水浴冷却的过程中,伴随着液-液相分离的结束,固-液相分离所形成的球晶结构会对液-液相分离起到一定的抑制作用,而且在20℃时的固-液分相速度相比于60℃时较快,过冷度的增大导致降温速率较快,20℃水浴冷却下的膜的球晶结构不够规整,球晶生长存在较大缺陷,使得膜孔孔径分布不均匀,表面粗糙;而60℃水浴冷却下的膜,由于经历相分离过程的时间充裕,缓慢结晶,晶核可以长成大晶粒,逐渐变大的球晶相互接触,成为连续体,球晶的边界逐渐消失,膜孔孔径分布相对均匀,表面相对平整.
2.4.2 凝固浴温度对膜性能的影响
表3所示为相同制膜条件及工艺下,仅改变凝固浴温度对膜的水通量、截留率和孔隙率的影响,其中,PVDF的浓度及复合稀释剂的比例恒定.
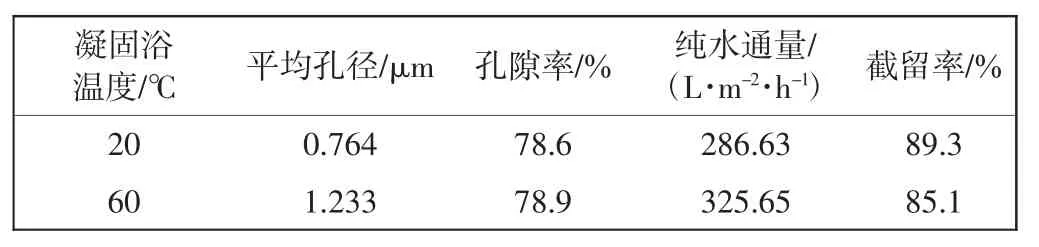
表3 凝固浴温度对膜性能的影响Tab.3 Effect of coagulation bath temperature on performance of membranes
由表3可知,60℃水浴冷却下膜的纯水通量和平均孔径较大,截留率较低,而膜的孔隙率几乎不变.利用TIPS法制膜的过程中,膜孔隙的产生除添加成孔剂外,还有一部分是在对膜内残余稀释剂进行萃取的过程中在膜内部及表面形成一定的缺陷从而发展成孔.因此,在聚合物浓度及稀释剂含量一定时,膜孔隙率变化不大,表明凝固浴的温度不影响膜的孔隙率.针对平均孔径及水通量的变化,可以解释为当凝固浴温度与铸膜液温度相差较小时,降温速率减慢,相分离过程持续的时间较长,结晶完善,形成的晶体尺寸较大,相应的膜孔孔径也会变大,水通量随之升高.
3 结论
本文将与PVDF相溶性良好的DBP以及相溶性较差的DOP按照一定比例混合作为复合稀释剂,通过低温热致相分离法制备得到PVDF平板膜,同时考察了PVDF的浓度、复合稀释剂的配比、降温速率以及凝固浴温度对膜结构与性能的影响.得出以下结论:
(1)由于非溶剂致相分离和热致相分离机理并存,故在膜结构中同时存在指状孔结构和海绵状孔结构;
(2)PVDF浓度的变化会极大地影响膜的微观结构.当PVDF处于低浓度时,膜为球粒堆积结构,随着浓度的增大,逐渐向双连续结构转变;并且随着复合稀释剂中DOP的含量增加,膜结构也会由最初的球粒堆积结构转变为双连续结构;
(3)膜的纯水通量和孔隙率均随着PVDF浓度的增大而减小;
(4)当PVDF质量分数为40%时,拉伸强度达到最大,为 1.80 MPa;
(5)选择20℃空气冷却作为体系的降温方式,纯水通量可达311.14 L/(m2·h),膜的平均孔径为0.972 μm;当凝固浴的温度达到60℃时,膜的纯水通量为325.65 L/(m2·h),平均孔径为1.233 μm;
(6)改变体系的降温方式以及凝固浴的温度对膜孔隙率的影响不大.
猜你喜欢
广州化工(2022年11期)2022-06-29
农业灾害研究(2022年1期)2022-05-07
粘接(2021年5期)2021-06-29
数学大王·中高年级(2021年4期)2021-04-27
环境卫生工程(2021年1期)2021-03-19
装备制造技术(2020年3期)2020-12-25
森林工程(2020年5期)2020-09-17
核化学与放射化学(2020年4期)2020-08-21
家庭影院技术(2019年8期)2019-08-27
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
