
克唑替尼治疗肺癌肝毒性与CYP3A5基因多态性的相关性分析
2018-11-08 02:56:42顾鹏王璇周月琴
中华肺部疾病杂志(电子版) 2018年5期
顾鹏 王璇 周月琴
肺癌是全球最常见的恶性肿瘤,其中85%的患者分型为非小细胞肺癌(non-small cell lung cancer, NSCLC)[1-2]。随着精准医学的发展,NSCLC的治疗已经进入个体化时代。2007年,科学家们发现了棘皮动物微管相关蛋白4-间变性淋巴瘤激酶(echinoderm microtubule associated protein-like 4/anaplastic lymphoma kinase, EML4-ALK)融合基因与肺癌相关,而针对EML4-ALK 融合基因靶点的小分子抑制剂克唑替尼在多项研究中被证实具有良好的临床疗效及耐受性[3-4]。
克唑替尼主要通过肝微粒体酶进行代谢,酶的代谢活性直接影响克唑替尼在体内的血药浓度[5]。CYP3A是药物代谢酶细胞色素P450(cytoehromeP450, CYP450)的一个亚家族,是参与药物代谢的主要酶系,主要分布在小肠和肝[6-7]。基因变异是CYP3A5酶表达的主要调控方式,CYP3A5被认为是造成个体间CYP3A活性差异的最主要原因[8]。CYP3A5 基因具有30多个单核苷酸多态性(single nucleotide polymorphisms, SNPs)位点,但并不是所有的基因突变均可引起酶活性改变。其中CYP3A5*3突变频率最高,携带突变型纯合子(CYP3A5*3/*3)的个体不表达CYP3A5,CYP3A5酶失去部分功能或丧失活性,导致对底物的代谢效率下降[9]。本文选择影响酶的催化活性的等位基因变异型——CYP3A5*3进行基因型研究,通过比较陆军军医大学(第三军医大学)新桥医院使用克唑替尼治疗肺癌患者的CYP3A5基因型差异,探讨克唑替尼相关的药物性肝损伤与CYP3A5基因多态性之间可能存在的相关性。
对象与方法
一、研究对象
选择2013年1月至2017年12月在陆军军医大学(第三军医大学)新桥医院采用克唑替尼治疗的84例NSCLC患者。其中男性36例,女性48例,中位年龄44.3岁,病理诊断4例为腺鳞癌,其他均为腺癌,12例为Ⅲb期,其他均为Ⅳ期。克唑替尼为一/二线治疗的各占32例,三线治疗20例,既往治疗方案包括紫杉醇联合铂类,多西他赛联合铂类,培美曲塞二钠联合铂类。以患者血清转氨酶或胆红素升高为表现肝毒性,分为肝毒性组32例、无肝毒性组52例,性别(男/女)分别为12/20和24/28,年龄分别为(46.85±12.34)岁和(43.98±11.61)岁,身高分别为(170.84±6.99)cm和(162.67±7.45)cm,体质量分别为(69.03±13.32)kg和(62.30±14.25)kg。两组患者年龄、性别、身高及体质量等比较,差异均无统计学意义(P>0.05)。
纳入标准: ①组织病理学或细胞学证据的Ⅲb期-Ⅳ期NSCLC(国际肺癌研究协会第7版NSCLC分期标准为据);②经Ventana IHC或荧光原位杂交技术(fluorescence insitu hybridization, FISH)或RT-PCR基因检测ALK重排融合基因阳性;③按标准剂量口服克唑替尼胶囊200~250 mg,2 次/d。排除标准:患者在服用克唑替尼之前有肝功能异常。
二、研究方法
收集患者的性别、年龄、身高、体质量等一般资料,病理类型及分期,是否肝转移,是否有肝炎病史,服用克唑替尼的剂量、时间,以及用药后监测肝功能的相关指标:丙氨酸氨基转移酶(alanine transaminase, ALT),门冬氨酸氨基转移酶(aspartate transaminase, AST),总胆红素(total bilirubin, TBIL)及碱性磷酸酶(alkaline phosphates, ALP)。肝毒性按常见不良反应事件评价标准(common terminology criteria for adverse events, CTCAE)5.0版标准进行评价。肝毒性分级:1级(血清转氨酶>正常值上限-3.0倍正常值上限;ALP>正常值上限-2.5倍正常值上限;TBIL>正常值上限-1.5倍正常值上限);2级(血清转氨酶>3.0~5.0倍正常值上限;ALP>2.5~5.0倍正常值上限;TBIL>1.5~3.0倍正常值上限);3级(血清转氨酶>5.0~20.0倍正常值上限;ALP>5.0~20.0倍正常值上限;TBIL>3.0~10.0倍正常值上限)。
抽取患者5 ml静脉血,用PHARM-GENE 200 SNP分析样本处理试剂(耀金分)进行DNA序列测定的分析样本处理,用实时荧光定量 PCR 仪,型号:RT-CyclerTM 436/TL988(博奥生物科技有限公司),进行SNP 基因型分析[10]。
三、统计学方法
采用SPSS 17.0 统计软件分析患者性别、年龄、身高、体重的组间差异,对无肝毒性对照样本的CYP3A5*3基因进行Hardy-Weinberg 平衡检验,α=0.05。应用STATA 10.0 软件进行非条件性二元Logistic 回归分析,校正年龄、性别,计算目标SNP 基因型的优势比(oddsratios,OR)和95%可信区间(confidence intervals,CI)。所有P值均为双侧检验。
结 果
一、肝功能损伤情况
84例患者中32例出现以血清转氨酶或胆红素或碱性磷酸酶升高为表现的肝毒性。肝毒性发生的中位时间为41 d(15~162 d)。发生1级肝功能损伤的有16例,发生2级肝功能损伤的有8例,发生3级肝功能损伤的有8例,没有患者发生4级肝功能损伤,见表1。其中有4例肝功损伤3级患者发生了INR值(国际标准化比值)的轻度升高。在发生2级和3级肝毒性的16例患者均有再次服用小剂量克唑替尼的经历,其中5例再次出现了克唑替尼导致的肝毒性,见表1。
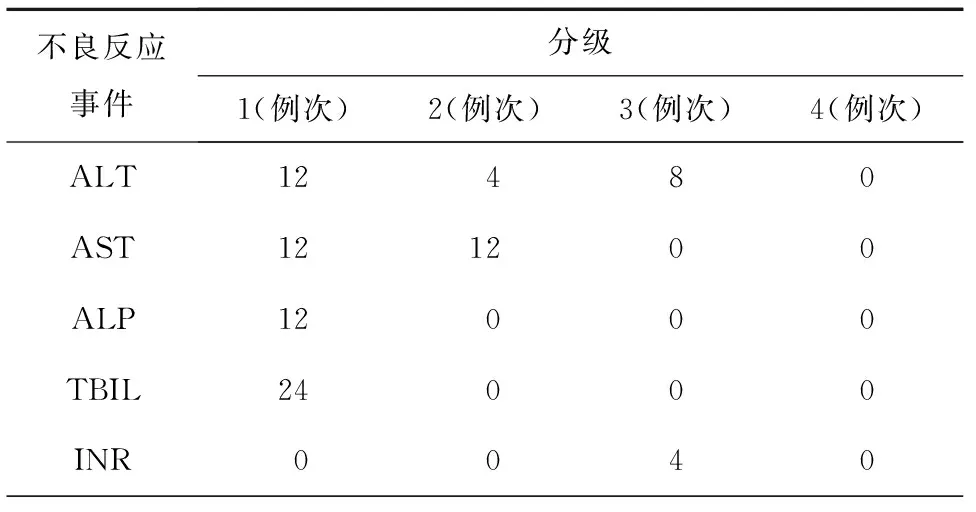
表1 肝功能损伤例数分布
注:ALT-丙氨酸氨基转移酶;AST-门冬氨酸氨基转移酶;ALP-碱性磷酸酶;TBIL-总胆红素;INR-国际标准化比值
二、CYP3A5多态性基因分型情况
不同种族间CYP3A5的变异率有所不同,其中CYP3A5*3 在白种人的突变频率为77.6%,在黑种人的突变频率为70.6%,而在中国人、日本人中的突变频率分别为71%~76%、71%~85%[11]。本实验中,CYP3A5*3的基因突变频率为78.57%,与文献报道相符。无肝毒性对照组通过了Hardy-Weinberg平衡检验(P=0.837),见表2。

表2 各组间CYP3A5*3基因型
注:GG-野生纯合型;GA-突变杂合型;AA-突变纯合型
三、相关性分析
经非条件性二元Logistic 回归分析,在定义的共显性和显性遗传模型下,发现CYP3A5*3突变与克唑替尼肝毒性的发生无显著性关联,见表3。
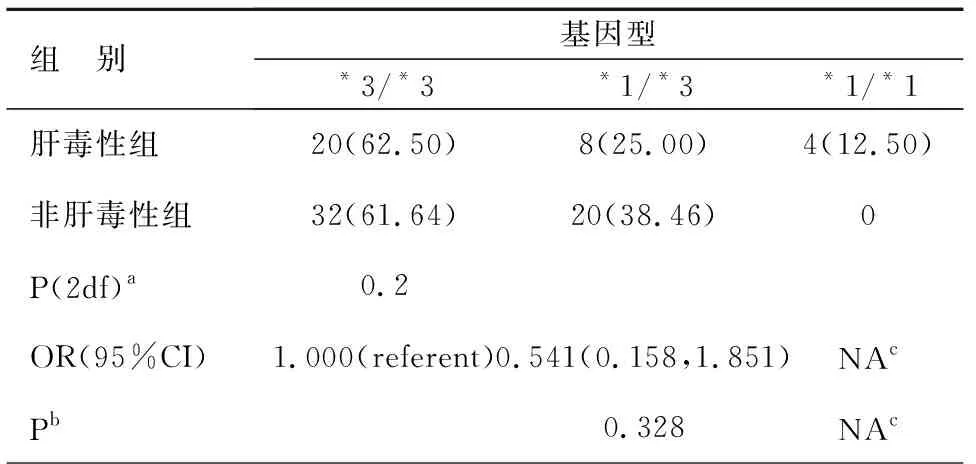
表3 CYP3A5*3基因型与克唑替尼肝毒性的
注:a-全局性P值(df=2):肝毒性组与非肝毒性组基因型频率比较的χ2检验;b-非条件Logistic回归分析P值(校正年龄和性别);c-由于缺少基因型数据无法进行计算

讨 论
抗肿瘤药物的研发与上市十分迅速,特别是针对信号通路分子靶点的抗肿瘤药物的开发已成为重点领域。与传统的细胞毒性抗肿瘤药物相比,以细胞信号转导分子为靶点的抗肿瘤药物具有更高的抑瘤效率和更低的毒副作用,但仍存在不同程度的脱靶效应。研究显示,在药物安全性评价中失败以及从药品市场上撤销的药物中,超过一半以上的原因是由肝脏毒性导致的,抗肿瘤药物的肝脏毒性值得关注。
酪氨酸激酶抑制剂是一类新型的靶点抗肿瘤药,其基本作用原理是通过与竞争性结合细胞外的配体结合位点,阻断酪氨酸激酶的活化,抑制受体的激活,从而达到抑制肿瘤的恶性增殖以及促进肿瘤细胞凋亡。随着上市后临床使用时间的延长以及患者人群的扩大,该类药物的毒性被逐渐报道,包括心脏毒性、骨髓毒性、肺毒性等等,其中肝脏毒性是该类药物不良反应率较高的一种。研究显示,达沙替尼等药物肝毒性报道率最高药物化学结构中,都存在“迈克尔反应受体结构”,能够共价结合并消耗细胞内谷胱甘肽,从而破坏肝脏的氧化应激体系。再者酪氨酸激酶类都是口服药物,胃肠道吸收后会经过肠系膜血管进入门静脉系统经过肝脏,在肝脏蓄积,以上结果无疑提示这些药物极易破坏肝细胞内的氧化还原的平衡,引起氧化应激,损伤肝细胞。
克唑替尼是酪氨酸激酶抑制剂的一种,在Ⅰ、Ⅱ、Ⅲ期临床试验中显示有10%~38%的患者出现转氨酶升高,中国患者较高加索人种转氨酶升高更多见[12-15]。酪氨酸激酶的肝脏毒性作用除了与细胞应激、免疫反应相关外,近年来也认为与药物代谢酶CYP450的基因多态性有关[16-18]。克唑替尼主要通过CYP3A的途径代谢,亦是CYP3A的抑制剂[19-23]。剂量依赖性细胞毒性机制和免疫过敏机制都在小分子化合物所致肝毒性中被提及[24-27]。然而,哪种机制占主导地位要取决于个体。前一种机制可能在服用小剂量克唑替尼安全的人群中占主导地位。后一种机制可能在服用小剂量克唑替尼仍然导致肝毒性的人群中占主导地位。不管是哪种机制,在肝内未代谢的激活型克唑替尼和其具有生物活性的代谢物,可能是其所致肝毒性的决定性因素。
CYP3A5基因多态性会对克唑替尼所致肝毒性有影响这种假设,在本研究中没有被证实,发现携带CYP3A5*1(表达CYP3A5)的患者与携带CYP3A5*3/*3(不表达CYP3A5)的患者其发生肝毒性的风险没有显著性差异。然而有趣的是,在本研究中,发生2级和3级肝毒性的患者中有6例同时服用了CYP3A4抑制剂,且接受克唑替尼的小剂量再次给药(250 mg,1次/d)后,具有*3/*3的5例再次发生了克唑替尼导致的肝毒性。即同时服用CYP3A4抑制剂且具有*3/*3的患者,在再次小剂量使用克唑替尼时都会发生肝毒性,这种肝毒性在服用CYP3A4抑制剂且不具有*3/*3的患者中没有发生。上述发现使本研究不能完全排除服用克唑替尼导致肝毒性与CYP3A5的活性降低有关。所以,如果患者因遗传因素导致了CYP3A5活性降低,在CYP3A4功能因联合用药而受到影响时,CYP3A5活性降低会使克唑替尼代谢不足,蓄积在体内,导致肝毒性。此外,大多数CYP3A4抑制剂都是P-糖蛋白抑制剂。在本研究的联合用药中,氨氯地平不是P-糖蛋白抑制剂,硝苯地平和地尔硫也不是P-糖蛋白抑制剂。这从一定程度上排除了P-糖蛋白对克唑替尼造成影响的可能性。
本研究仍然有一些不足。因为这是一项回顾性分析,部分患者在入组时已经停止使用克唑替尼,所以没有药动学/药效学的研究。另外,由于肝损伤患者进行肝穿刺活检较少,没有通过检测肝活检标本来对药物在肝脏内的浓度和CYP酶的活性进行测定。克唑替尼所致肝毒性的机制期望在将来的前瞻性研究中进一步明确。CYP3A5活性降低虽然不能直接预示克唑替尼肝毒性的发生,但存在一定相关性。克唑替尼个体代谢不同,包括联合用药和免疫过敏机制对克唑替尼和/或其代谢物的影响,这些复杂的因素综合导致了其肝毒性的产生[28-31]。在CYP3A5活性降低的患者中,应尽量避免使用CYP3A4抑制剂。相信更多的研究将会进一步阐明克唑替尼肝毒性的机制,使患者能更好的避免药物相关不良反应的发生,使临床用药更加合理、安全、有效。
猜你喜欢
中国临床新医学(2023年9期)2023-10-18 07:14:24
实用药物与临床(2022年12期)2023-01-17 01:01:40
临床肺科杂志(2020年7期)2020-07-02 09:21:34
中国医药导报(2019年5期)2019-04-28 10:29:32
装备制造技术(2017年10期)2017-12-28 09:11:00
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
现代检验医学杂志(2015年1期)2015-02-06 01:59:07
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
中国癌症杂志(2015年6期)2015-01-04 13:26:33
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
