
荧光素酶表达载体构建及其在巴斯德毕赤酵母的表达
2018-07-11 06:41罗展浩吴清平张菊梅丁郁李程思潘力吴慧清
现代食品科技 2018年6期
罗展浩,吴清平,张菊梅,丁郁,李程思,潘力,吴慧清
(1.华南理工大学生物科学与工程学院,广东广州 510006)(2.广东省微生物研究所,省部共建华南应用微生物国家重点实验室,广东省菌种保藏与应用重点实验室,广东省微生物应用新技术公共实验室,广东广州 510070)(3.暨南大学理工学院食品科学与工程系,广东广州 510632)
萤火虫荧光素酶(Firefly Luciferase,FL)能在O2、Mg2+、ATP存在条件下,催化D-荧光素转化为氧合荧光素并发出蓝绿荧光(550~580 nm)[1]。ATP生物发光反应产生的荧光强度在一定范围内与 ATP的量成正比,通过一定阶段恒定的ATP的量可以反映细菌等微生物的数量,该方法与传统的细菌培养菌落计数法有良好的相关性[2],由于ATP生物发光具有高灵敏度、检测简便等优点,该方法已广泛应用于食品工业各个领域。此前本实验室在荧光素酶的活体提取[3]、荧光素的化学合成[4]和微生物的生物发光快速检测试剂性质研究[5]上进行较为系统的探索,开发了基于ATP生物发光的微生物快速检测盒。面对食品生产过程微生物污染的问题,基于ATP生物发光的微生物快速检测盒的研发愈发重要,因此通过不同表达系统大量快速生产荧光素酶可迎合市场需求。
1985年,De Wet[6]首次克隆北美萤火虫荧光素酶,并在大肠杆菌内表达,随后各种荧光素酶被克隆和鉴定[1]。不少研究学者选择大肠杆菌作为表达系统[7~9],以研究酶的生产和应用。但是通过大肠杆菌表达蛋白需要抗生素添加以保持遗传性和存在着大量表达时会产生包涵体等问题。戎晶晶等[10]发现荧光素酶在大肠杆菌BL21(DE3)中表达形式主要为包涵体,夏蕾等[11]采用大肠杆菌 M15表达可溶蛋白,比活达到1.43×109RLU/mg,但产量只有0.68 mg/30 mL,对于大规模生产相当不利。
近年来由于荧光素酶的高灵敏度,单纯作为报告基因,在真核系统中的表达如在裂殖酵母和毕赤酵母[12]、酿酒酵母[13,14]表达也有过报道,但是作为外源蛋白在毕赤酵母内的表达和纯化以及作为 ATP检测试剂进行测试评估和应用却未见报道。毕赤酵母作为一种常用的高效生产蛋白的真核表达系统,相比大肠杆菌具有遗传稳定、可进行高密度培养、具有蛋白翻译后糖基化修饰,蛋白能正确折叠,能进行目标蛋白的定向转运如胞外分泌等一系列优点[15]。
本研究将北美萤火虫(Photinus pyralis)荧光素酶基因luc克隆到毕赤酵母表达载体pPIC9K,并进行毕赤酵母的稳定转化。在得到阳性转化子的基础上进行甲醇诱导发酵,成功表达了荧光素酶。本研究首次实现了荧光素酶在毕赤酵母GS115体内的稳定表达、纯化及酶活鉴定的初步研究。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
巴斯德毕赤酵母菌株GS115(P. pastorisGS115)和大肠杆菌DH5α(Escherichia coliDH5α)为本实验室保存,质粒 pPIC9K购自 Invitrogen公司,pGL2-control购自Promega公司。
1.1.2 主要试剂和培养基
Pfu DNA聚合酶、AvrII、NotI、SacI限制酶、T4 DNA连接酶、Page Ruler prestained protein ladder购自赛默飞世尔科技公司;DNA marker、氨苄青霉素、遗传霉素(G418)、购自上海生工生物工程公司;鼠抗荧光素酶抗体购自Santa Cruz Biotechnology公司;羊抗鼠IgG抗体购自全式金公司;质粒提取试剂盒、普通DNA胶回收试剂盒、普通DNA纯化试剂盒均购自天根生化科技公司;DEAE Sepharose FF层析柱、Superdex 75pg购自纯原生物公司;超滤离心管、luminol购自Merck Millipore公司;D-荧光素为本实验室制备;其他试剂为国产分析纯试剂。
LB培养基、YPD培养基、MD培养基、MM培养基、BMMY培养基、BMGY培养基按照Invitrogen公司的毕赤酵母表达手册制备。
1.1.3 仪器设备
蛋白纯化仪 AKTA start,GE Healthcare公司;Multiporator多功能电转移电融合仪,Eppendorf公司;Milli-Q纯水机,Millipore;Avanti J-26S XP高速冷冻离心机,Beckman公司;Centrifuge 5424小型离心机,Eppendorf公司;Nexus GSX1梯度PCR仪,eppendorf公司;VC-505超声波细胞破碎仪,Sonics公司;Chemidoc MP全能型成像系统,Bio-rad公司;Glomax 20/20 luminometer荧光检测仪,Promega公司。
1.1.4 引物
本文所用的引物如表1所示。
1.2 方法
1.2.1 北美萤火虫荧光素酶基因luc的克隆与表达载体的构建
从NCBI(GenBank:X65324.2)获取北美萤火虫荧光素酶基因序列luc,设计PCR引物见上表1,由华大基因合成。以pGL2-control为模板,引物luc-F、luc-R,PCR扩增获得荧光素酶全长基因luc。反应条件:96 ℃ 10 min;96 ℃ 40 s,60 ℃ 40 s,72 ℃ 2 min,35个循环;72 ℃ 8 min。PCR产物纯化后,与质粒pPIC9K分别进行双酶切,产物回收后用T4连接酶进行连接。连接产物热击转化感受态大肠杆菌E.coliDH5α,感受态细胞制备及转化方法参考《分子克隆实验指南》[16]进行。转化后在 LB固体培养基(Amp终浓度为100 μg/mL),挑选阳性转化子过夜培养后提取质粒进行酶切验证,并送华大基因公司测序。构建正确的质粒命名为pPIC9K-luc。

表1 荧光素酶基因扩增与验证的PCR引物Table 1 PCR primers for luciferase gene amplification and confirmation
1.2.2 重组菌 GS115/pPIC9K-luc的构建及转化子的筛选
重组质粒pPIC9K-luc经SacI线性化后,用普通DNA纯化试剂盒纯化,电转化P.pastorisGS115感受态,转化产物加入1 mol/L冰冷山梨醇,低速振荡培养1 h后,涂布MD选择性固体培养基,30 ℃培养至单菌落出现。酵母感受态细胞制备参考文献[17]。
将单菌落转移到含0.5 mg/mL、1 mg/mL、2 mg/mL遗传霉素G418的YPD固体培养基,30 ℃培养3~6 d,筛选能在高浓度遗传霉素G418的YPD培养基上生长的单菌落。单菌落转接到YPD液体培养基培养1 d,用煮-冻-煮[18]方法提取基因组作为扩增模板进行PCR验证,引物为α-factor和3’AOX1,反应条件同上。同样的方法将空质粒pPIC9K转化GS115感受态细胞作为对照。
1.2.3 重组菌P.pastorisGS115/pPIC9K-luc的Mut+、MutS表型鉴定
将筛选所得的转化子均影印到MD和MM培养基,同时能在两种培养基中良好快速生长的为 Mut+表型;在前者快速生长但在MM培养基中生长缓慢的为MutS。
1.2.4 重组P.pastorisGS115/pPIC9K-luc甲醇诱导表达
挑取重组的P.pastoris单菌落接种到10 mL液体BMGY培养基中,30 ℃、200 r/min振荡培养约24 h作为种子液。取1 mL种子液接种于50 mL BMGY培养基培养24 h至OD600约为3~6。4 ℃,5000 r/min离心5 min,沉淀重悬于50 mL BMMY培养基(含2%甲醇),进行甲醇诱导表达,20 ℃、200 r/min,每隔24 h补加甲醇至终浓度为2%,同时每12 h取样,直接测量发酵液的酶活性,96 h后最终收获发酵液。
1.2.5 发酵粗酶液的提取
50 mL发酵液5000 r/min 离心5 min得到上清粗酶液Ⅰ,沉淀用 2 mL荧光素酶溶解液(0.2 mol/L Tris-HCl,5 mmol/L Mg2+,1 mmol/L EDTA,pH 7.8)冲洗,离心后得到上清粗酶液Ⅱ。菌体沉淀纯水洗涤数次后加入 2 mL酵母裂解缓冲液(0.2 mol/L Tris-HCl,1 mmol/L PMSF,5 mmol/L EDTA,10%甘油,pH 8.0),加入直径为425 μm~600 μm 玻璃珠进行涡旋破碎12次(30 s涡旋,30 s冰上放置为1次)后,冰浴超声破碎8 min(5 son,5 soff),12000 r/min离心10 min,得到胞内酶清液Ⅲ。
1.2.6 重组菌 GS115/pPIC9K-luc表达产物的SDS-聚丙烯酰胺凝胶电泳和western blotting鉴定
三种粗酶液Ⅰ、Ⅱ、Ⅲ,加入SDS proteinloading buffer沸水浴5 min,短暂离心作电泳上样样品。同时GS115/pPIC9K全菌体作为阴性对照,进行SDS-聚丙烯酰胺凝胶电泳(5%浓缩胶,12%分离胶)分析。电泳过后,电转至PVDF膜,5%脱脂奶粉4 ℃过夜封闭,分别用鼠抗荧光素酶抗体作为一抗孵育40 min,以辣根过氧化氢酶标记的羊抗鼠 IgG抗体为二抗孵育 20 min后,H2O2与luminol混合为反应底物显影,进行蛋白印迹分析。
1.2.7 粗酶液的酶活测定
20 μL甘氨酸缓冲液(GDB)(25 mmol/L甘氨酰甘氨酸二肽,5 mmol/L Mg2+,1 mmol/L EDTA-Na2,0.4%α-环糊精,0.15%二乙氨乙基葡聚糖,pH 7.2),10 μL 1 mmol/L D-荧光素,10 μL 1 mmol/L ATP 溶液组成的反应液,加入10 μL粗酶液开始生物发光反应,立即放入荧光检测仪中,integrate time 4 s模式测定荧光强度室温下测定,发光强度以荧光检测仪的读数值-相对发光单位RLU(Relative Light Units)表示酶活。
若测试粗酶液本底荧光发光强度,将反应体系中的10 μL ATP溶液换成超纯水,以评估细胞残留ATP对发光的影响程度。
1.2.8 粗酶液中蛋白的纯化
50 mL诱导表达96 h的培养液,离心取沉淀,加入2 mL酵母裂解缓冲液,玻璃珠与超声破碎后,离心取上清液作为粗酶液。粗酶液用超滤离心管(MWCO 50 ku)进行超滤并且更换缓冲液NTE(20 mmol/L NaHCO3,10 mmol/L Tris,1 mmol/L EDTA,pH 7.8)。蛋白浓缩液经DEAE-FF阴离子交换柱进行第二步纯化,用含0~1 mol/L NaCl的NTE缓冲液进行梯度洗脱,收集蛋白峰。后经Superdex 75 pg分子筛凝胶层析第三步纯化,NTE洗脱收集蛋白峰。最后用SDS-PAGE检验纯化效果,并对相应粗酶液进行酶活鉴定。
1.2.9 ATP浓度生物发光检测
将酶稀释到 0.1~0.5 mg/mL,采用如下体系进行ATP 生物发光检测:20 μL GDB,20 μL 酶液,5 μL 1 mmol/L荧光素作为反应体系。分别加入 5 μL 1×10-6~1×10-12mol/L ATP 后,立即放入荧光检测仪中,integrate time 4 s模式测定荧光强度,室温下进行测定。取发光强度的对数和 ATP浓度的对数作荧光发光值-ATP浓度的ATP标准曲线。
取1×10-6~1×10-10mol/L范围内的浓度已知的ATP样品作为待测ATP样品,根据标准曲线估计待测样品ATP浓度,并对检测体系进行评价。
1.2.10 数据统计分析
所有酶活性测定均重复3次,结果以平均值和标准偏差表示,图表采用Excel 2010和Origin 8.0数据处理软件加工处理。
2 结果与讨论
2.1 表达载体的构建
2.1.1 荧光素酶基因的克隆与验证
根据方法1.2.1构建的重组表达载体pPIC9K-luc如图 1(a),荧光素酶基因luc插入到 5’AOX1与3’AOX1(TT)之间的表达框内。用NotI与AvrII双酶切pPIC9K-luc验证转化子,酶切结果如图1(b),得到两条明显条带:荧光素酶基因luc(1653 bp)与线性化质粒pPIC9K(9273 bp),测序结果也验证荧光素酶基因序列正确,表达载体pPIC9K-luc构建成功。
2.1.2 重组菌的构建及转化子的筛选
限制酶SacI酶切得到线性化的重组载体pPIC9K-luc,电转化山梨醇制备的毕赤酵母感受态细胞P. pastorisGS115,在缺乏组氨酸的MD筛选平板上初筛一批转化子41个,并且进一步复筛,挑选了最高能在含1 mg/mL G418的YPD平板上生长的菌落2个。G418筛选浓度的高低反映着整合到染色体组目的基因的拷贝数目,剂量效应被认为影响异源蛋白在毕赤酵母表达系统中最重要的因素,基因拷贝数越高,表达量越大[19]。本文未能筛选到抗2 mg/mL G418的高拷贝转化子,较大可能因为初筛的转化子数目远不足,只有41个,一般His+转化子中自发多拷贝整合的概率为1~10%。

图1 重组质粒pPIC9K-luc的构建(a)和重组质粒pPIC9K-luc的酶切鉴定(b)Fig.1 Construction of recombinant plasmid pPIC9K-luc (a) and gel electrophoresis of pPIC9K-luc digested with restriction enzymes(b)
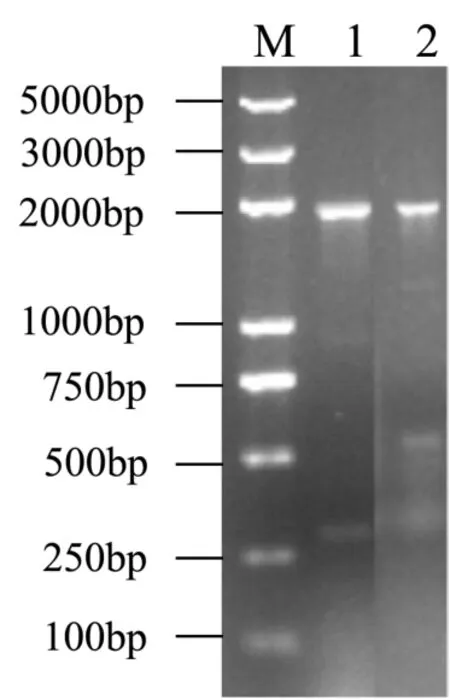
图2 重组酵母GS115/pPIC9K-luc的PCR验证Fig.2 PCR identification of recombinant P. pastoris GS115/pPIC9K-luc
本文复筛得到的抗1 mg/mL G418的转化子,进一步提取的酵母染色体组DNA,进行PCR验证如图2所示。出现接近2 kb的条带,与α-factor和荧光素酶融合的基因片段(约1.9 kb)相吻合,证明重组载体的目的基因成功重组插入到核染色体组。筛选得到的 2个菌株编号为 GS115/pPIC9K-1,GS115/pPIC9K-2,经验证均为Mut+表型。同时空载体转化的酵母命名为GS115/pPIC9K。
2.2 荧光素酶的诱导表达与纯化
2.2.1 发酵液与上清液的酶活初步测定
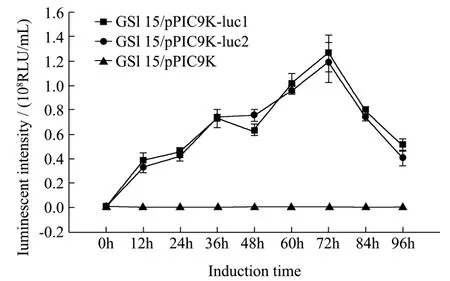
图3 发酵液酶活测定Fig.3 The enzyme activity assay of fermentation broth
每12 h取样保存于-20 ℃冰箱的发酵液样品作为粗酶液,取10 μL加入到荧光素酶发光反应液中进行活性测定,发光强度如图3所示,发酵时间在72 h内,两株 GS115/pPIC9K-luc的发酵液的酶活性呈增长趋势,荧光素酶逐渐累积。在72 h时,GS115/pPIC9K-luc1发酵的酶淸液达到峰值为1.25×108RLU/mL。随着发酵时间延长,发酵液酶活迅速降低,96 h仅剩下40.7%最高酶活:5.09×107RLU/mL。这种酶活迅速降低的原因可能是蛋白酶水解所致。该结果也为日后的优化实验选取最佳发酵时间提供了参考。GS115/pPIC9K-luc2菌株的发酵所得酶酶活变化趋势和每个时间点的绝对酶活与菌株1相差不大。
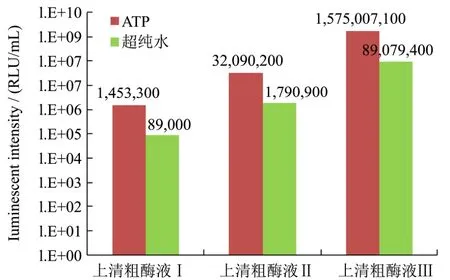
图4 上清粗酶液酶活测定Fig.4 The enzyme activity assay of supernatants
诱导表达96 h最终收获的发酵液,经过离心、洗涤沉淀、破碎细胞再离心后得到3份上清粗酶液Ⅰ、Ⅱ、Ⅲ,同样进行酶活测定。如图4所示,发酵液上清即粗酶液Ⅰ的发光活性较低为1.45×106RLU/mL;上清粗酶液Ⅱ的酶活约为前者的 20倍,为 3.21×107RLU/mL;酵母裂解的胞内粗酶液Ⅲ的酶活最高,达到1.58×109RLU/mL。这组结果表明酵母表达的荧光素酶没有完全释放到胞外,大部分保留细胞内。
本研究构建了毕赤酵母-荧光素酶表达株,期望在α因子分泌肽的引导下进行胞外表达荧光素酶[20],但绝大多数荧光素酶未能有效分泌到胞外。与Yasuo等[21]在烟草植物真核细胞表达FL,只在胞内发现酶发光活性的结果一致。某些研究[22~24]表明,荧光素酶羧基端的丝氨酸-赖氨酸-亮氨酸三肽(SKL过氧化物酶体定位序列),能够将其转运到过氧化物酶体中,导致荧光素酶分泌胞外分泌量较低。但胞外淸液粗酶活也达到1.45×106RLU/mL,表明荧光素酶转运到过氧化物酶体的途径可能被过表达的荧光素酶饱和,而逃逸进入外泌途径。有报道证明,发酵过程添加酸水解酪蛋白能够有效增加蛋白的表达量[25],表面活性剂如吐温-20的添加能显著增加蛋白的分泌量[26],这些方面都为提高荧光素酶表达量和促进胞外分泌提供可能,也指明了后续研究工作的方向。三种粗酶液的酶活测试试验中,测试体系不外加ATP,以纯水代替,观察粗酶液中残留的细胞ATP对发光强度产生的影响,通过本底发光值表示。测试体系外加ATP的发光强度作为非本底值,发现前者为后者的5%~6%,表明发酵液和胞内破碎粗酶液仍然含有一定量酵母细胞产生的ATP残留,对表达蛋白的酶活测定造成一定影响,在处理上作相应扣除以反映酶液的真实酶活水平。
2.2.2 重组菌 GS115/pPIC9K-luc表达产物SDS-PAGE电泳以及western blot分析

图5 重组菌GS115/pPIC9K-luc表达产物SDS-PAGE电泳分析Fig.5 SDS-PAGE analysis of protein expressed by recombinant GS115/pPIC9K-luc
将上清粗酶液Ⅰ、Ⅱ、Ⅲ进行SDS-聚丙烯酰胺凝胶电泳,结果如图5所示。结合western blot分析,发现表达的荧光素酶分子量略大于70 ku(高于理论预测大小62 ku),且大部分存在于细胞内而非胞外。在大肠杆菌内表达的荧光素酶均为60 ku[8],推测可能有潜在糖基化发生。用 NetNGlyc 1.0Server(http://www.cbs.dtu.dk/services/NetNGlyc/)对重组蛋白潜在糖基化位点进行预测,发现3个潜在的N-糖基化位点,导致酶实际分子量较其理论分子量大。糖基化作为毕赤酵母系统最常见的蛋白翻译后加工形式,不同位点的糖基化发挥不同作用[27]。文献报道显示,N-糖基化能显著提高酶的稳定性、分泌性和活性[28]。Yang[29]在毕赤酵母表达的中华根霉脂肪酶时发现,部分位点糖基化可提高分泌性,部分可提高酶的活性和稳定性。Chang[30]通过表达特定糖基化位点缺失的烟曲霉木聚糖酶,发现糖基化的缺失导致酶热稳定性显著降低。同理,荧光素酶的糖基化亦为提高其热稳定性和酶活性提供了可能,对提高荧光素酶的可利用程度是有帮助的SDS-PAGE分析中上清粗酶液Ⅲ的荧光素酶蛋白条带较为明显,而上清粗酶液Ⅰ没有显示相应条带,这与三份样品的酶活分析结果相一致,再次证明表达的蛋白大部分存在于细胞内。

图6 重组菌GS115/pPIC9K-luc表达产物蛋白印迹分析Fig.6 Western blotting analysis of protein expressed by recombinant GS115/pPIC9K-luc
Western blot结果如图6所示,胞外、胞内粗酶存在其他微弱的杂带,空白对照不存在任何条带出现,而泳道4更在30 ku出现较深杂带,这表示蛋白受到不同程度的降解,造成降解片段与抗体的结合显现条带,与图3所示结果吻合。研究表明[31~34],低温诱导蛋白表达利于提高蛋白稳定性,降低蛋白酶浓度与活力,增加细胞的存活率。20 ℃诱导时,AOX1酶及与甲醇诱导过程中能量代谢途径相关酶的活性比 30 ℃诱导时的都要高[35]。因此选用 20 ℃作为诱导温度,一方面能够较好地利用碳源,另一方面尽可能降低蛋白酶酶活,提高酶活性表达。前期实验工作确实发现30 ℃表达的胞外荧光素酶酶活只有20 ℃的5%~17%。20 ℃的发酵温度有一定的表达优势,但结果表明20 ℃还不足以完全抑制蛋白酶的水解作用。
2.2.3 荧光素酶的纯化

图7 纯化产物的SDS-PAGE分析Fig.7 SDS-PAGE analysis for each step of purified products
50 mL菌液培养所得的菌体,直接破碎得到粗酶液经过超滤、DEAE阴离子交换层析和分子筛凝胶层析,得到2.4 mg蛋白,其比活达到7.0×108RLU/mg,纯化倍数达到19.3,如表2,与夏蕾[8]报道的在大肠杆菌纯化的比活为1.43×109RLU/mg,纯化倍数为10.6,比活上并不突出,但是在酶的回收上有着优势,未来可通过改变发酵条件和高密度发酵技术,在大规模生产上取得突破。如图7所示,纯化后的荧光素酶的电泳图出现两条大小略大于70 ku的条带,为不同程度糖基化的荧光素酶。萤火虫荧光素酶作为一种来源于真核细胞的蛋白,适度的糖基化更接近于萤火虫体内的存在形式,对酶活稳定存在一定作用。因此在毕赤酵母中表达糖基化的酶是大肠杆菌系统无法实现的,针对该系统表达的荧光素酶,未来可以就糖基化对荧光素酶所产生的效应作进一步的研究。
Verma[12]曾在比较真核启动子的活性中,使用AOX1启动子在裂殖酵母表达荧光素酶,Leskinen[36]、Ainsworth[37]也同样就酿酒酵母系统中启动子活性检测的研究中使用过荧光素酶,但均没有在纯化和活性上作过多的研究。本研究初次在毕赤酵母中以荧光素酶的异源表达纯化、应用为目的,获得的荧光素酶达到预期的产量和活性。

表2 不同纯化步骤后重组酵母表达的荧光素酶的特性分析Table 2 The characteristics of luciferase expressed by recombinant P.pastoris after different purification steps
2.3 ATP浓度生物发光检测
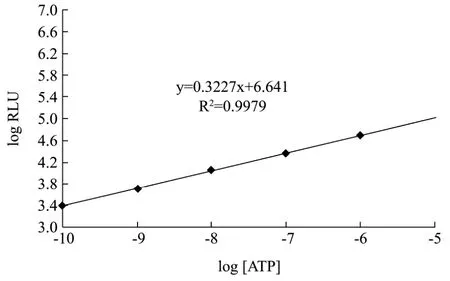
图8 ATP生物发光标准曲线Fig.8 Standard curve of ATP assay
采用本研究的荧光素酶进行ATP生物发光反应,绘制 ATP标准曲线,发现本研究的酶在 10-6~10-10mol/L存在良好线性关系,y=0.3227x+0.641,R2=0.9979(图 8)。选取线性范围内的三个浓度 ATP,配制成ATP溶液作为待测样品,进行ATP浓度检测来评价酶的实用效果。具体ATP测量结果如下表3。

表3 纯化后荧光素酶对ATP样品的生物发光检测Table 3 Biological luminescence detection for ATP solution sample by purified luciferase
上述结果表明,纯化的荧光素酶能够较好地检测ATP并反映其真实浓度。
3 结论
3.1 荧光素酶的一个重要应用就是通过检测微量ATP从而对微生物进行定量检测,与传统的琼脂平板培养法相比,有着快速、简便、高灵敏度、可自动化操作等优点[38],如果采用免疫磁珠对样品进行前处理,使用含有荧光素酶的微生物ATP快速检测盒,则可对食源性致病微生物进行检测,从而能够监测、预防食品加工过程食源性致病微生物的污染。
3.2 相比起大肠杆菌表达系统,毕赤酵母有着其独特的优势:外源基因的遗传性能稳定、可对产物进行糖基化修饰,能够进行高密度培养发酵。本研究构建了荧光素酶的表达载体pPIC9K-luc,运用毕赤酵母表达系统,成功表达荧光素酶。但是未能使酶进行有效的分泌,诱导表达 96 h后胞内酶活达到了 1.58×109RLU/mL。对胞内酶进行了有效的阴离子层析和分子筛凝胶层析纯化,获得单一的荧光素酶,比活为7.0×108RLU/mg,纯化倍数达到19.3倍,产量为48mg/L。
3.3 本研究利用真核表达系统毕赤酵母来表达荧光素酶,为商业化生产荧光素酶提供了理论依据。未来可通过过氧化氢酶体定位序列的缺失和发酵条件的优化,增加异源表达荧光素酶的胞外分泌量以简化下游纯化步骤,使其更加适应酶的工业化生产要求。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
中日友好医院学报(2021年1期)2021-04-14
山东医药(2020年9期)2020-05-20
癌症进展(2018年11期)2018-12-30
天然产物研究与开发(2018年4期)2018-05-07
中成药(2018年1期)2018-02-02
现代园艺(2017年19期)2018-01-19
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
中国医药导报(2015年27期)2015-02-28
