
大鼠双下肢骨折合并失血对心肌细胞损伤的作用及其机制
2018-06-08 01:27
中国应用生理学杂志 2018年2期
李朝晖,张 颖,路 艳,陆庆明,谢晓华△
(解放军总医院南楼综合外科,北京100853)
据WHO统计,全世界每年仅交通事故伤害就导致超过120万人死亡[1]。严重的创伤失血(traumatic hemorrhage)是可以直接威胁生命的危急重症,也是中青年人重要的致死原因。当机体受到严重创伤时,常因血容量丢失导致有效循环血量减少,引起严重微循环障碍,引发一系列延续性局部/全身创伤反应,造成脏器功能不可逆损伤,包括心肌组织,但其确切发生机制尚不清楚。目前已知,在心肌损伤的发生过程中,心肌细胞凋亡具有重要意义。本文通过在体观察及体外心肌细胞培养研究,探讨双下肢骨折创伤失血反应可否诱导心肌细胞发生凋亡反应,以为下一步深入研究肢体创伤出血导致心肌损伤的发生机制奠定基础。
1 材料与方法
1.1 大鼠双下肢骨折创伤失血模型的建立与分组
健康雄性SD大鼠20只,体重250~300 g(购自军事医学科学院动物中心),常规饲养条件下正常饮食饮水。大鼠随机分为对照组和骨折创伤组(n=10)。腹腔注射3%异戊巴比妥钠(80 mg/kg体重)麻醉大鼠,使用自制重物坠落装置致大鼠双后肢股骨干粉碎性骨折。方法为:将大鼠固定在装置底盘上,将重物升至固定高度后任其自由坠落,分别砸伤大鼠双侧股骨干致其闭合性粉碎性骨折,随后尾动脉抽血约10ml,1 h后双倍液体(生理盐水)回输,包扎,固定[2]。全部大鼠分别在创伤发生后 0、1、2、4、8、12、16、24及 48 h尾动脉取血,并于 48 h时处死大鼠,留取左室前壁心肌标本,液氮保存备用。
1.2 心肌细胞损伤模型
取正常大鼠心脏,经Langendorff装置灌注,从灌流液中分离心肌细胞,置CO2培养箱中悬浮培养24 h后,加入创伤血清继续培养48 h,构建心肌损伤模型。
1.3 血清炎症相关细胞因子的测定
在创伤后的不同时间点(创伤前和创伤后1、2、4、8、12、16、24及 48 h),分别取各组大鼠尾动脉血液,2 000~3 000 r/min离心 20 min,取血清,以备ELISA法测定。
1.4 心肌组织HE染色
心肌组织取材、固定、脱水、石蜡包埋、切片,常规苏木素-伊红染色,中性树胶封固。
1.5 心肌细胞凋亡的测定
取心肌细胞制备切片,灭活内源性过氧化物,生物素标记后,采用TUNEL法测定。
1.6 凋亡调控基因bcl-2/bax测定
取30mg心肌碾碎后,加入1 ml Trizol振摇,离心 12 000 r/min×15min(4℃),采用 RT-PCR法测定基因表达。引物序列为:baxForward 5’-3’TCGTCCATCGAGGATGACTTC, Reverse 5’-3’AACACCACAATTAAGGCAGGG;bcl-2Forward 5’-3’ACGTGGACCTCATGGAGTG,Reverse 5’-3’TGTGTATAGCAATCCCAGGCA
1.7 凋亡调控基因 bcl-2/bax蛋白表达的测定(Western blot)
取心肌碾磨,加入RIPA 300μl裂解30 min,4℃12 000 r/min离心,SDS-PAGE,电转液中反应后转膜,加入一抗后4℃孵育过夜,给予二抗结合反应,经DAB显色,测定Bcl-2/Bax蛋白的表达量。
1.8 统计学处理
计量数据以均数±标准差()表示,所有资料均运用GraphPad Prism医学统计软件进行检验,两组等方差数据之间的比较采用成组t检验。
2 结果
2.1 大鼠双下肢骨折创伤失血对心肌损伤的影响
2.1.1 心肌组织HE染色 阅片发现,骨折创伤失血反应会导致心肌组织发生一定程度的损伤,心肌细胞形态发生改变,排列不规则,肿胀明显,伴有大量巨噬细胞聚集(图1,见彩图页Ⅳ)。
2.1.2 心肌细胞培养TUNEL染色 实验表明,加入创伤血清进行心肌细胞培养后,能够复制创伤模型,创伤组培养的心肌细胞中出现大量核染成棕褐色的细胞,此即为凋亡的心肌细胞(图2,见彩图页Ⅳ)。对照组凋亡指数为3.65±0.81,创伤组凋亡指数为 27.47±1.83,两组有明显差异(P<0.01)。
2.2 大鼠双下肢骨折创伤失血对血清炎性因子含量的影响
检测大鼠骨折创伤后不同时间点血清炎性因子含量的变化,发现在创伤后短时期(8 h)内血清IL-2水平急剧下降,8 h降至最低值(74.22±8.90 pg/ml,n=6),较对照组(214.41±11.99 pg/ml,n=10)显著减低 (P<0.05);随着时间的延长,血清IL-2的水平开始缓慢增加,一直到创伤后48 h大鼠血清IL-2水平仍显著低于对照组水平(P<0.05)。
在骨折失血后,创伤组大鼠血清IL-6、IL-10水平开始急剧升高,创伤后4 h达到峰值,分别为(455.70±14.31pg/ml,n=6)和(189.71±8.05 pg/ml,n=6),较对照组(分别为 203.75±4.98 pg/ml,n=10和 64.52±8.36 pg/ml,n=10)显著增加(P<0.05,P<0.05)。随着时间的延长,血清 IL-6、IL-10水平呈逐步下降趋势,创伤后16~24 h恢复至对照组水平。
在骨折创伤失血后的早期,创伤组TNF-α表达水平迅速升高,创伤后1 h即可达到最高值(58.22±3.51 pg/ml,n=6),较对照组(45.24±5.94 pg/ml,n=10)显著升高(P<0.05),随后即呈快速下降趋势(图 3)。
2.3 双下肢骨折创伤失血对大鼠心肌bcl-2/bax表达的影响
骨折创伤发生后,Western blot方法检测显示,心肌bax蛋白表达量增高,而bcl-2蛋白表达量下调。
RT-PCR检查结果则提示,对照组bax基因相对表达量为(0.98±0.03,n=10),创伤组为(3.96±0.21,n=6),两组差异显著(P<0.01)。而创伤组bcl-2基因相对表达量为(0.50±0.08,n=6),较之对照组(1.01±0.05,n=10)明显下调(P<0.01,图 4)。

Fig.3 Changes in contentofserum associated inflammatory factors in rats
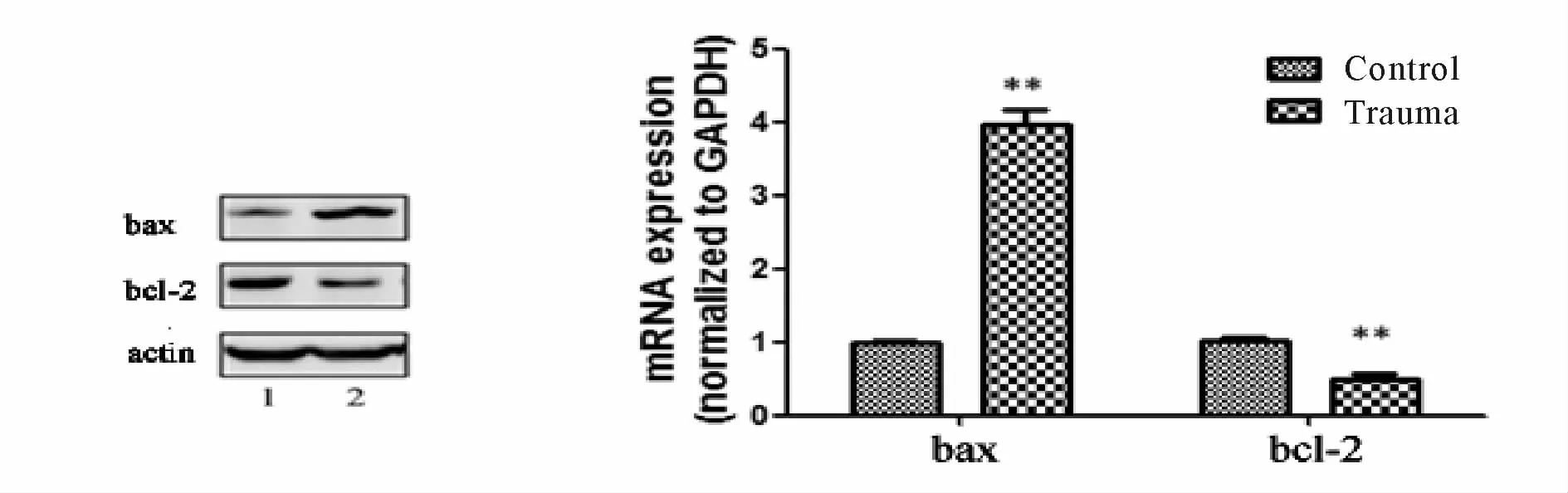
Fig.4 Changes of bax/bcl-2 expression in ratmyocardial tissue 1:Control group;2:Trauma group
2.4 心肌细胞培养测定凋亡调控基因bax/bcl-2表达的变化
Western blot检测发现,加入创伤血清培养的损伤心肌细胞中,bcl-2蛋白表达量减少,而bax蛋白表达量增加。
进一步以 RT-PCR方法检测基因 bax/bcl-2表达。结果显示:对照组心肌细胞中促凋亡基因bax的相对表达量为(0.99±0.09),而创伤组为(3.95±0.28),两者差异显著(P<0.01)。而对照组抑制凋亡基因 bcl-2的相对表达量为(0.99±0.08),创伤组为(0.56±0.05),二者相差具有明显统计学意义(P<0.05,图 5)。

Fig.5 Changes of bax/bcl-2 expression in cultured cardiomyocytes
3 讨论
目前认为,创伤所导致的心肌损伤机制,与炎性因子介导的心肌细胞凋亡、氧化应激损伤、能量代谢障碍、钙超载等诸多因素有关,这些因素之间还存在相互链接,交织成复杂的网络。本研究通过ELISA检测也发现,创伤后 IL-2、IL-6、IL-10、TNF-α均呈现时间依赖性规律的变化,首先是TNF-α在创伤后极短时间内达峰值(1 h内),然后缓慢下降;随即IL-6、IL-10升高,4 h左右达到峰值,然后逐步下降;而IL-2呈现相反的改变,创伤后急剧下降,4~8 h时降至谷底,随后缓慢回升。这样的变化规律,与其他学者的研究报导是一致的[3]。这说明创伤可使机体产生和释放一系列炎症相关因子,通过级联放大产生“全身炎症瀑布效应”,造成心肌受损。如IL-6和TNF-α,可引起机体多种炎性介质的释放失调[4],TNF-α表达水平的持续增高既可直接导致心肌细胞膜受损、破坏,同时还可以募集其他炎性细胞浸润,直至心肌发生坏死、纤维化。而IL-6则可以通过氧化应激机制促使心肌细胞发生凋亡[5],TNF-α还可以增加IL-6的表达,产生协同作用,加速心肌细胞损伤。IL-10通过信号转导机制直接/间接降低凋亡消音蛋白的活性,上调机体对创伤损害的敏感性阈值,避免凋亡发生[6]。
HE染色发现,大鼠双下肢骨折创伤失血反应可以导致心肌不同程度受损,心肌细胞肿大,炎性细胞浸润。而且,培养心肌细胞TUNEL染色证实,创伤可以诱导心肌细胞发生凋亡。对于心肌细胞凋亡的认知曾有争议。曾有人认为,心肌细胞是一种最终分化的细胞,其死亡过程应为坏死,一旦坏死发生则不可再生。但随着凋亡相关研究的逐步深入,心肌细胞凋亡这一现象得到普遍的认同。心肌缺血、动脉粥样硬化、心肌病变等心血管疾患的不断发展均可以导致心肌细胞凋亡,进而加重心脏功能受损;反之,心肌细胞凋亡又可以加速心脏疾患的进程,累及心功能不断下降。资料表明,压力过负荷所致心衰过程中,常常伴有心肌细胞肥大,而细胞数量减少,凋亡系这种量减少过程的主要原因;即使是极小量的减少,也极可能会明显恶化心力衰竭的进程。甚至有研究提示,单一细胞死亡,通过与邻近细胞间信号转导,也会对整个心脏功能产生影响[7]。体外心肌细胞培养观察到,凋亡在短时间内(2 h)即可发生[8];而在体研究表明,凋亡的发生可能需要4~24 h[9,10],甚至更长。相对于某一固定的时间点,所测得的凋亡反应仅能反映相对固定的短时间内细胞的凋亡进程,只有历经足够长的时间过程检测,才可能符合机体心脏的实际状况[11]。
心肌细胞凋亡过程中,Bcl-2/Bax是一对最有效也是最关键的凋亡蛋白,具有非常重要的意义。Bax分布广泛,细胞膜、细胞浆及细胞核中都有它的定位,如在线粒体外膜、内质网、核周膜等处[12]。在各种凋亡诱发信号介导下,游离于胞浆中的Bax发生迁移,与Bcl-2异源寡聚化为Bax-Bcl-2,可以使Bcl-2的活性受到抑制;同时还可以直接同源寡聚化为Bax-Bax,促进Cyt-c释放入胞浆,松解凋亡激活因子Apaf-1与 Bcl-2的耦合,进而触发凋亡[13]。与之相反,Bcl-2的主要作用是抑制心肌细胞凋亡,是公认的长命基因,通过C末端插入域定位于线粒体外膜、内质网、核周膜等处,能够封闭Bax激活的mPTP孔道,进而阻断凋亡[14]。研究表明,细胞内 Bcl-2/Bax的比例变化,对于是否产生凋亡倾向具有关键意义,直接反映心肌受损程度[15]。一般情况下,Bcl-2/Bax处于稳定的动态平衡状态,一旦机体受到刺激,通过信号传递打破这种动态平衡,将产生截然相反的结局。因此,Bax/Bcl-2的比例可以作为检测凋亡的一个很重要的指标,如出现Bax表达增加而Bcl-2表达下调,则促进凋亡;反之,则可抑制凋亡反应。有资料显示,心肌缺血初始阶段,p38MAPK及其下游通道活化,促进Bax易位至线粒体,进而启动凋亡;这一过程涉及凋亡激活因子Apaf-1,以及死亡蛋白(caspase-9)的易化过程;Bcl-2通过维持线粒体膜的完整性,阻断该反应过程[16,17]。
综上,本研究表明,大鼠双下肢骨折创伤失血反应,无论在体内或体外,均可以诱导凋亡调控基因Bcl-2/Bax表达比例发生变化,呈现促凋亡发生样改变,导致远隔脏器的损伤。通过一系列细胞因子介导,最终可以引发心肌细胞凋亡,造成更加严重的伤害,但其确切传递机制尚需进一步探讨。针对性地阻断其传导通路,或上调其保护性应答机制,可能会为临床提高危重创伤的救治、避免心肌损伤的发生提供一定的帮助。
[1] Wang ZG.Road traffic injuries[J].JMed Col PLA,2005,20(1):1-3.
[2] Michael F,Hagen A,Christian Z,etal.Experimental traumamodels:an update[J].J Biomed Biotechnol,2011,2011(1):636-650.
[3] Ayala A,Perrin MM,Meldrum DR,et al.Hemorrhage induces an increase in serum TNF which is not associated with elevated levels of endotoxin[J].Cytokine,1990,2(3):170-174.
[4] 赵自刚,张 静,牛春雨.多器官功能障碍综合征发病机制的某些研究进展[J].微循环学杂志,2004,14(3):80-83.
[5] Pudil R,Pidrman V,Krejsek J,et al.The effect of trimetazidine on c-reactive protein,cytokines and adhesion molecules in the course ofacutemyocardial infarction[J].Acta Medica,2001,44(4):135-140.
[6] GrilliM,Barbieri I,Basudev H,et al.Interleukin-10modulates neuronal threshold of vulnerability to ischaemic damage[J].Eur JNeurosci,2000,12(7):2265-2272.
[7] Teiger E,Than VD,Richard L,etal.Apoptosis in pressure overload induced hearthypertrophy in the rat[J].JClin Invest,1996,97(12):2891-2897.
[8] Oberhammer FA,Pavelka M,Sharma S,et al.Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor(beta)1[J].PNAS,1992,89(12):5408-5412.
[9] Kang PM,Izumo S.Apoptosis and heart failure:a critical review of the literature[J].Circ Res,2000,86(11):1107-1113.
[10]Lim H,Fallavollita JA,Hard R,et al.Profound apoptosismediated regionalmyocyte loss and compensatory hypertrophy in pigswith hibernatingmyocardium[J].Circulation,1999,100(23):2380-2386.
[11]Zhao ZQ,Velez DA,Wang NP,et al.Progressively developed myocardial apoptotic cell death during late phase of reperfusion[J].Apoptosis,2001,6(4):279-290.
[12]Barclay LA,Wales TE,Garner TP,etal.Inhibition of proapoptotic Bax by a noncanonical interaction mechanism[J].Mol Cell,2015,57(5):873-886.
[13]Hoetelmans R,van Slooten HJ,Keijzer R,et al.Bcl-2 and Bax proteins are present in interphase nuclei ofmammalian cells[J].Cell Death Differ,2000,7(4):384-392.
[14]Chang SF,Sun YY,Yang LY,etal.Bcl-2 gene family expression in the brain of rat offspring after gestational and lactational dioxin exposure[J].Ann NYAcad Sci,2005,1042:471-480.
[15] Prince PS,Dhanasekar K,Rajakumar S.Preventive effects of vanillic acid on lipids,bax,bcl-2 and myocardial infarct size on isoproterenol-inducedmyocardial infarcted rats:a biochemical and in vitro study.[J].Cardiovasc Toxicol,2011,11(1):58-66.
[16]Capano M,Crompton M.Bax translocates tomitochondria of heart cells during simulated ischaemia:involvement of AMP-activated and p38 mitogen-activated protein kinases[J].Biochem J,2006,395(1):57-64.
[17]Wang X,Welsh N.Bcl-2maintains themitochondrialmembrane potential,but fails to affect production of reactive oxy-gen species and endoplasmic reticulum stress,in sodium palmitate-induced beta-cell death[J].Ups J Med Sci,2014,119(4):306-315.
猜你喜欢
中国现代医生(2022年21期)2022-08-22
中国现代医生(2022年21期)2022-08-22
世界科学技术-中医药现代化(2022年2期)2022-05-25
当代医药论丛(2021年24期)2022-01-20
世界科学技术-中医药现代化(2021年7期)2021-11-04
中华养生保健(2020年9期)2021-01-18
科学咨询(2020年10期)2020-04-01
中成药(2017年8期)2017-11-22
中国医药科学(2017年9期)2017-08-04
放射学实践(2016年6期)2016-12-15
