
基于Discovery Studio分子模拟药物设计软件的拉伸分子动力学
2018-06-02 03:23张心苑毛雪石
智慧健康 2018年7期
张心苑,毛雪石
(北京协和医学院中国医学科学院药物研究所,北京 100050)
0 引言
Discovery Studio (简称DS)是基于Windows/Linux系统,面向药学领域的新一代分子建模和模拟环境,它应用于药物结构功能研究以及药物发现[1]。Discovery Studio研究领域包括:疾病的发病机理研究、新药发现和设计、计算药物化学、肿瘤药物研究等[2]。
Discovery Studio功能模块包括基于结构的药物发现和设计模块、基于片段的药物设计模块、基于药效团的药物发现和设计模块、基于小分子的药物发现和设计模块蛋白质建模及模拟模块、分子力学、分子动力学、分子力学/量子力学模拟模块等。Discovery Studio 2017新增拉伸分子动力学功能,在分子动力学的基础上引入拉伸力场,能够更好地分析配体(药物分子)、受体的结合方式,本文主要介绍分子动力学模块的拉伸分子动力学功能。
拉伸分子动力学研究的主要问题是利用拉伸分子动力学计算配体(药物分子)结合受体的自由能、研究配体(药物分子)的解离过程、研究蛋白质的去折叠过程或构象变化、拉伸离子或配体(药物分子)通过通道蛋白、构建平均力势(PMF)。蛋白质类别(结构)不同,展开不同结构的蛋白质所需的拉伸力、温度不一样,拉伸分子动力学过程(由高温和外力诱导的去折叠过程)有差异[3]。不同蛋白质的拉伸分子动力学过程的差异行为与蛋白质折叠的拓扑结构和蛋白质的二级结构元素的稳定性有关。
1 建立并运行拉伸分子动力学模拟
本文以1TEN、1TIT的晶体结构为例,在Discovery Studio中载入1TEN、1TIT的pdb文件。其中,1TEN是一种纤维粘连蛋白,1TIT是一种免疫球蛋白,两种蛋白的二级结构都是β折叠、β转角[4-6]。
1.1 选择一对原子作为拉伸方向
用rNc表示蛋白质一级结构(氨基酸序列)中N端(NH2-)中氮原子到C端(—COOH)中碳原子连线所形成的向量。拉伸力的方向平行于rNc并且力的方向是使N端(NH2-)中氮原子到C端(—COOH)中碳原子远离的。
首先,确定N端及C端氨基酸,右键单击molecule window出现display style,选择protein,再选择color by,选择N-to-C Terminal,出现的3D结构视图显示只有蓝、红两部分。在蓝色部分内的氨基酸就是N端的氨基酸,在红色部分内的氨基酸就是C端的氨基酸。N-C端向量即为拉伸方向,蛋白质的结构特点不同确定的拉伸方向不同。
1.2 设置拉伸动力学参数,建立拉伸动力学模拟
1.2.1 确定运算时间
如果恒力足够大,蛋白质拉伸运算完成的时间与合适的力拉伸运算完成的时间不同[7]。运算时间的确定与施加力的大小有关,需要调节拉伸力的同时观察拉伸分子动力学运算时间是否合适。从标准分子动力学运算结果可看出1TIT蛋白质中运算时间的平衡点在2400ps后,因此将运算时间设为3000ps最为合适。
1.2.2 确定运算温度
最佳的拉伸分子动力学温度为450K,在300K-450K的温度范围内都合适,温度为500K时,拉伸时间小于500ps,拉伸速率较快,不适合观察拉伸分子动力学过程中蛋白质构象的变化[8]。
1.2.3 确定运算拉伸力
拉伸力等于150pN时可以达到70-250Å的拉伸距离,无法完全拉伸蛋白质[9]。拉伸力小于等于250pN时无法完全拉伸蛋白质,拉伸力大于400pN时拉伸速度过快,不适于观察蛋白质结构的变化,最佳拉伸力为400pN。
1.2.4 运行拉伸分子动力学模拟
运算拉伸动力学后出现600个conformation(构象),蛋白质完全拉伸后的分子构象如图1所示。

图1 拉伸方向图(左) 拉伸分子动力学运算结果图(右)
2 拉伸动力学结果分析
2.1 观察不同时间点、不同拉伸距离的蛋白质结构状态
拉伸动力学方法可以提供整个过程中原子水平的细节,从轨迹中获得一些快照,可表示拉伸动力学过程中的构象变化。现分别在rNC=43.26Å,rNC=56.99Å,rNC=64.02Å,rNC=71.72Å,rNC=79.96Å,rNC=87.15Å,rNC=94.83Å,rNC=102. 57Å 及初始状态和完全拉伸状态按照时间节点将8幅图显示如图2。(1 Å=10-10m,Å主要用于原子物理、晶体学)。
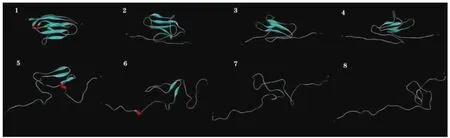
图2 不同时间点的轨迹图
2.2 绘制拉伸距离随时间变化图,观察拉伸动力学的平衡点
观察拉伸分子动力学模拟结果,从模拟的第一个构象一直到最后一个构象的变化动画,可以发现距离量是一直变化的,拉伸距离的变化范围为43.26 Å-102.57 Å。
分析拉伸距离随时间变化的原因是找到拉伸平衡状态,最佳状态时的拉伸距离随时间变化图应该是逐渐上升趋势增加到一定值时趋于平衡的状态[10]。若增加拉伸模拟时间或增加拉伸力可以更快地达到平衡状态,做出含平衡状态的拉伸距离随时间变化图。
2.3 绘制RMSD,编译动力学轨迹监控数据
RMSD图表示蛋白质结构差异,趋势逐渐增大至稳定,如图3所示。Tools explorer选项卡中选择Analyze Trajectory,再选择 Analyze,Create Plot设置RMSD参数:Properties选择Distance1,Analysis type选择RMSD、RMSF、Properties,其他参数按默认设置即可。
2.4 根据多条运算轨迹计算自由能
根据拉伸分子动力学运算结果适当调整拉伸力、拉伸时间、拉伸温度的值,会得到若干条运算轨迹,根据30-40条轨迹可以准备地计算自由能,现根据一条拉伸分子动力学计算自由能。Tools explorer选项卡中展开Run Simulations,在Steered Molecular Dynamics菜单下点击Calculate Free Energy from Steering Force,得到计算自由能随拉伸距离变化的图4。
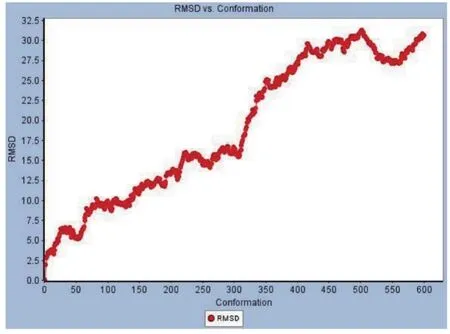
图3 RMSD图

图4 自由能随拉伸距离的变化图
3 结论
在Discovery Studio 2017软件中新增拉伸分子动力学的计算功能,通过软件模拟拉伸分子动力学过程体现蛋白质的去折叠过程中的构象变化、配体(药物分子)的结合及解离过程,进一步研究并优化药物分子与受体蛋白的结合方式。Discovery Studio 2017软件的拉伸分子动力学模块有强大的分析动力学运算结果的工具,能够有效分析药物分子在力场中与靶标蛋白的结合方式。拉伸分子动力学的计算功能还有利于基于靶标的药物设计及药物的高通量筛选,在模拟大分子药物、化学、生物系统中具有重要作用。
[1] 李亚梅,廖端芳.应用Discovery Studio软件预测烟酸姜黄素酯抗动脉粥样硬化的作用靶点[J].中国医药导报,2013,10(17):16-18.
[2] Paci E,Karplus M.Unfolding Proteins by External Forces and Temperature:The Importance of Topology and Energetics[J].Proceedings of the National Academy of Sciences of the United States of America,2000,97(12):6521-6526.
[3] 王浩,徐利楠,孙玉娜,等.Ydj1p二聚体中β14~β15与domain-Ⅲ分离的拉伸分子动力学模拟研究[J].生物信息学,2013,11(3):167-171.
[4] Wang Y,Cai W S,Chen L,et al.Molecular dynamics simulation reveals how phosphorylation of tyrosine 26 of phosphoglycerate mutase 1 upregulates glycolysis and promotes tumor growth[J].Oncotarget,2011,8(7).
[5] 季海涛,张万年,周有骏,等.药物化学计算机辅助教学方法的建立与应用[J].中国医学教育技术,2002,16(3):169-170.
[6] Paci E,Caflisch A,Plückthun A,et al.Forces and energetics of hapten-antibody dissociation: a biased molecular dynamics simulation study[J].Journal of Molecular Biology,2001,314(3):589-605.
[7] Boyce,S. E.;Mobley D. L.;Rocklin G.J.;et al. Predicting Ligand Binding Affinity with Alchemical Free EnergyMethodsin a Polar Model Binding Site[J].J.Mol.Biol,2009,394,747-763.
[8] 周雷,徐利楠,周小红,等.Hsc70-Auxilin复合物的拉伸分子动力学模拟[J].生物信息学, 2014(2):110-116.
[9] Bavi N,Bavi O,Vossoughi M,et al.Nanomechanical properties of MscL alpha helices: A steered molecular dynamics study[J].Channels,2016,11(3).
[10] Park M S.Molecular Dynamics Simulations of the Human Glucose Transporter GLUT1[J].Plos One,2015,10(4):e0125361.
猜你喜欢
空气动力学学报(2022年4期)2022-08-23
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
黑龙江大学自然科学学报(2022年1期)2022-03-29
无机化学学报(2020年7期)2020-07-20
数学小灵通(1-2年级)(2020年6期)2020-06-24
中学生数理化·八年级数学人教版(2017年2期)2017-03-25
科技创新导报(2016年30期)2017-03-15
浙江大学学报(工学版)(2015年2期)2015-05-30
火炸药学报(2014年1期)2014-03-20
中国科技信息(2012年11期)2012-10-26
