
溃疡性结肠炎合并神经纤维瘤病1型:1例报道并文献复习
2018-05-30 08:22伍东升李全晓钱家鸣
胃肠病学和肝病学杂志 2018年5期
宋 晓,伍东升,黄 璨,吕 红,李全晓,李 骥,钱家鸣
中国医学科学院 北京协和医学院 北京协和医院 1.急诊科;2.消化内科;3.内科;4.临床医学系,北京 100730
病例患者,男,31岁,因“腹泻、黏液脓血便5年,加重半年”入院。患者于2012年7月出现腹泻,为黏液脓血便,10次/d,50 ml/次,伴里急后重,伴脐周绞痛,便后腹痛缓解。粪便常规示大量白细胞和红细胞(3~5)/HPF,阳性。结肠镜可见全结肠、直肠广泛性病变,表现为不规则浅溃疡,病理示结肠黏膜急慢性炎,见淋巴滤泡、隐窝脓肿。诊断为溃疡性结肠炎(ulcerative colitis, UC),予柳氮磺吡啶(1 g,qid)、泼尼松龙(40 mg,qd)治疗,泼尼松龙至2013年4月规律减停,仅口服柳氮磺吡啶(1 g,qid)维持。治疗6个月后患者大便为成形软便,少量黏液脓血,2~3次/d。2016年末上述症状复发,伴腹痛、骶髂关节疼痛。2017年1月就诊于我院查血常规正常,肝肾功能正常,超敏CRP 65.61 mg/L,血沉 60 mm/h,结肠镜示左半结肠、直肠可见多发息肉,息肉周围黏膜大致正常,息肉表面充血水肿明显,FICE染色表面腺管粗大呈松塔样疏松结构,部分肿物表面有白色糜烂,部分糜烂面可见不规则网状血管(见图1)。病理示黏膜急慢性炎,隐窝结构紊乱,可见隐窝脓肿。考虑UC复发,UC相关脊柱关节炎,予足量泼尼松(45 mg,qd)及柳氮磺吡啶(1 g,qd)治疗,症状缓解。半年后泼尼松逐渐减量至7.5 mg,柳氮磺吡啶(1 g,qid)直至本次入院(2017年9月6日)。复查肠镜:横结肠近脾曲至直肠可见节段性多发成簇分布息肉,形态同前,息肉基底部可见明显淋巴管扩张样改变,息肉之间的结肠黏膜正常。超声内镜下,可见息肉样的病变为低回声,黏膜下层结构完整。切除2枚较大息肉,病理诊断为黏膜显急性及慢性炎,伴肉芽组织形成,符合炎性息肉(见图2)。免疫组化:AE1/AE3(-),CD117(-),CD34(血管+),S-100(散在+),DOG-1(-),Desmin(-),SMA(-),IgG4染色(-)。既往:出生时即有躯干部、腋窝、双手雀斑及牛奶咖啡斑(见图3);20岁左右开始出现胸腹背部多发皮下结节,大者2 cm,触之柔软,我院皮肤结节活检病理为:真皮内肿瘤细胞团块,由纤细的梭形细胞组成,可见肥大细胞(见图4)。
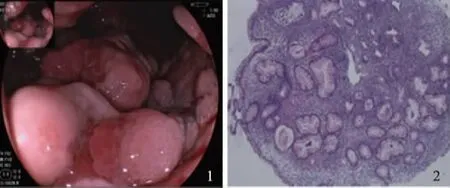
图1肠镜示:直肠多发大小不一息肉,表面色红,个别息肉可见纤维帽;图2息肉组织病理学改变符合炎性息肉改变(HE100×)
Fig1Colonoscopy:multiplerectalpolypsinsizes,thesurfacecolorwasred,fibercapcouldbeseenonindividualpolyps;Fig2Polymorphismofpolypsisconsistentwithchangesininflammatorypolyps(HE100×)
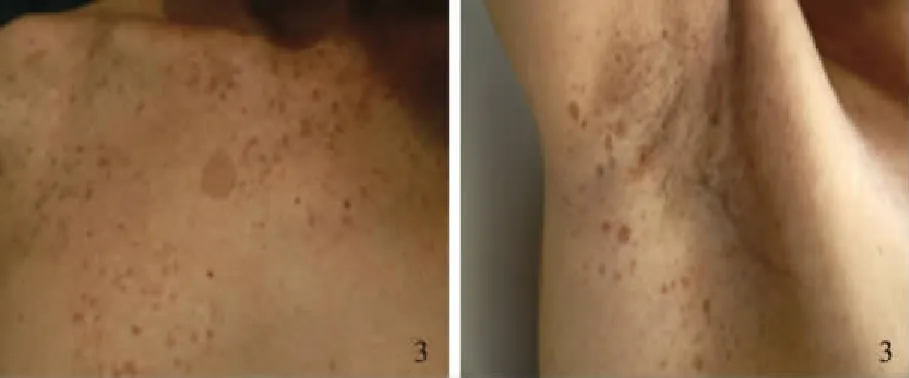
图3 躯干部牛奶咖啡斑、腋窝雀斑Fig 3 Flat pigmented lesions of the skin called café au lait spots on the trunk, and freckling of the axillae or regions
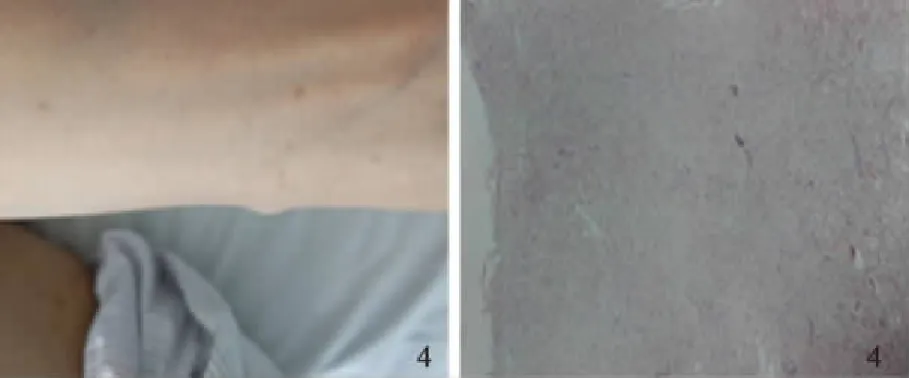
图4 皮下结节及皮肤结节活检符合神经纤维瘤改变Fig 4 Dermal neurofibroma and the biopsy of dermal neuro-fibroma conforms to neurofibromatosis
讨论本患者为青年男性,慢性病程,以腹泻、黏液脓血便起病,病程中出现双侧骶髂关节炎,肠镜示全结肠、直肠连续性病变,可见多发浅表性溃疡,活检病理可见隐窝脓肿,柳氮磺吡啶及激素治疗后病情缓解,综上,UC诊断明确。近期评估肠镜可见结直肠多发息肉,病理符合炎性息肉。值得一提的是,患者同时罹患神经纤维瘤1型(neurofibromatosis type 1, NF1),经PubMed及万方医学数据库进行文献检索,发现两者为共患病,本例报道为全球第5例报道,国内的首例报道。
神经纤维瘤病是周围神经系统的一种良性神经鞘肿瘤,临床和遗传学上有3种主要不同类型:NF1、NF2和施旺细胞瘤病。NF1又称von Recklinghausen病,是最常见的类型,是常染色体显性遗传病,可表现为家族聚集,也可散发突变。NF1基因是位于常染色体17q11.2的抑癌基因,NF1基因编码的神经纤维瘤蛋白(neurofibromin)是三磷酸鸟苷水解酶(guanosine triphosphate hydrolase, GTPase)活化蛋白家族的一员,NF1基因突变后,神经纤维瘤蛋白表达水平下降导致RAS信号通路的持续激活,从而刺激细胞生长和增殖[1]。病理上神经纤维瘤由施旺细胞、成纤维细胞、脱颗粒的肥大细胞和血管细胞组成。美国国立卫生研究院共识会议于1997年更新的NF1诊断标准是:(1)6个或以上在青春期前直径>5 mm或在青春期后直径>15 mm的咖啡牛奶斑;(2)2个或以上任何类型的神经纤维瘤或1个丛状神经纤维瘤;(3)腋窝或腹股沟区雀斑;(4)视胶质瘤;(5)2个或以上Lisch结节(虹膜错构瘤);(6)特征性骨病变,如蝶骨发育不良或长骨皮质增厚伴或不伴假关节;(7)有一级亲属(父母、同胞或子女)根据上述标准被诊断为NF1;满足以上标准中的两个标准,即可确诊为NF1[2-3]。该患者满足第1、2、3、5条,故可诊断为NF1。
NF1合并UC极其罕见。国外报道NF1的发病率为1/3 000~1/4 500[4-5],UC的发病率为21/100 000,若将二者视为无发病相关性的共患病,则同一例患者同时合并两种疾病的概率约为1/15 000 000[6]。因为病例数报道尚少,无法获取UC合并NF1患者与其他UC患者在诊治上的明显差异。在两种疾病发病机制方面的探讨中,一方面,肥大细胞在神经纤维瘤形成中存在作用,NF1患者的神经生长因子(NGF)水平升高,引发肥大细胞脱颗粒和释放炎症介质[7-10]。另一方面,UC患者脱颗粒的肥大细胞数量增加,有学者提出肥大细胞可能参与UC的发生,特别是在调节与神经元结构相关的炎症中发挥作用[11-12]。在已报道的4例病例中有2例报道结肠黏膜CD117表达增加,而CD117是KIT基因编码的酪氨酸激酶蛋白受体,肥大细胞及消化道Cajal间质细胞均表达CD117[11-12]。BARATELLI等[13]发现,他们所报道的病例中,显微镜下表现和免疫组织化学染色均显示结肠黏膜中与施旺细胞成分相关的肥大细胞显著增加,印证了肥大细胞参与了UC与NF1发病的假说。
在该患者的诊治中,其结直肠息肉增生十分明显,已知UC患者及NF1患者均可以出现消化道息肉样病变,那两者的肠道息肉有何不同吗?此前的4例UC合并NF1的个案报道中,有3例合并结肠息肉。其中,1例为结肠孤立性、中等大小、非出血性息肉,结肠黏膜CD117表达明显增加[14]。1例为距肛缘30 cm孤立性假性息肉,病理为炎性假性息肉[15]。1例为全结肠多发无蒂息肉,病理为假性息肉,固有层中CD117表达明显增加,神经元特异性烯醇化酶(NSE)和S100阳性提示存在施旺细胞成分[13]。1例无肠息肉[16]。
NF1较少累及胃肠道,可表现为弥漫的神经纤维瘤病/神经节瘤病,单个/丛状神经纤维瘤,壶腹周围良性肿瘤,和胃肠道间质瘤(gastrointestinal stromal tumour, GIST)。GIST是NF1最常见的胃肠道表现[17-18]。NF1患者出现胃肠道息肉样病变,通常表现为局部或弥漫性黏膜或黏膜下的神经纤维瘤/神经纤维瘤病,和/或神经节细胞瘤[19-20],而炎性/增生性息肉是否为NF1的明确肠道表现,目前尚无定论。ABBAS等总结了1949-2014年所发表的共15例NF1相关的肠道息肉案例,7例为单发息肉,8例为≥2个息肉,最常见的部位为结肠(6例),也见于胃、食管、小肠,直肠未见息肉,息肉直径为2~50 mm(平均20 mm)。病理上:大多数NF1患者的息肉,显示出炎性或幼年性息肉的特征:隐窝紊乱,黏蛋白细胞增生,黏蛋白滞留囊肿,浅表性溃疡,富含成纤维细胞样梭形细胞的炎症明显的固有层,明显的血管病变和嗜酸粒细胞浸润。AGAIMY等[21]认为,NF1患者肠道息肉的组织学表现多种多样,从以炎症为主,到纤维血管肉芽组织样为主和幼年样息肉为主等多种模式,建议把这些息肉分类为炎性息肉更好。2017年GOTO等[22]报道1例NF1相关的肠道息肉,为降结肠的2 cm孤立息肉,病理可见大量炎细胞浸润,黏膜和黏膜下血管病变,作者也认为该息肉形成与NF1密切相关。
UC发生炎性息肉的发生率各报道差异较大,可达4%~74%[23-24],2000年后发表的文献中,报道的发生率多为20%~40%[25]。发生炎性息肉与UC发病年龄、病程、活动程度无明显相关性,但目前普遍认为,重度活动和增生愈合过程更易形成炎性息肉,与结肠累及范围也呈正相关。UC炎性息肉最常见的形成部位依次为横结肠、降结肠、乙状结肠、直肠(特别是上段1/3),表现多样,呈弥漫性或局部分布,可为无蒂、有蒂、叶状,常大小不一[26]。
因此,UC肠道息肉与NF1息肉样病变有一定的相似性,组织学上均可表现为黏膜和/或黏膜下炎细胞浸润(包括肥大细胞)、纤维血管肉芽组织形成,修复后期可见上皮细胞增生;均无特征性的免疫组化染色。当然也存在不同:UC息肉多为多发性,以结直肠为主,黏膜下血管增生不明显;而NF1息肉样病变可单发也可多发,目前尚无直肠受累报道,可有显著的黏膜下血管增生。该患者的息肉为多发、大小0.3~3.0 cm,病理符合炎性息肉改变,且未见显著的血管病变、黏膜下层成分,且以乙状结肠、直肠病变尤著,更倾向于炎性息肉,为UC的修复期改变;但患者息肉病变不能完全除外NF1的因素参与。
本例报道是国内首例NF1与UC共患的报道。临床医师应增加对该共患疾病的认识,有助于更好地认识两者的发病机制,给予更合理的治疗。
[1] WIESEN A, DAVIDOFF S, SIDERIDIS K, et al. Neurofibroma in the colon [J]. J Clin Gastroenterol, 2006, 40(1): 85-86. DOI: 10.1097/01.mcg.0000190777.42656.19.
[2] GUTMANN DH D H, AYLSWORTH A, CAREY J C, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2 [J]. JAMA, 1997, 278(1): 51. DOI: 10.1097/00006534-199810000-00135.
[3] Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference [J]. Arch Neurol, 1988, 45(5): 575.
[4] EVANS D G, HOWARD E, GIBLIN C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service [J]. Am J Med Genet A, 2010, 152A(2): 327-332. DOI: 10.1002/ajmg.a.33139.
[5] LAMMERT M, FRIEDMAN J M, KLUWE L, et al. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment [J]. Arch Dermatol, 2005, 141(1): 71-74. DOI: 10.1001/archderm.141.1.71.
[6] ELLINGHAUS D, BETHUNE J, PETERSEN B S, et al. The genetics of Crohn’s disease and ulcerative colitis--status quo and beyond [J]. Scand J Gastroenterol, 2015, 50(1): 13-23. DOI: 10.3109/00365521.2014.990507.
[7] STASER K, YANG F C, CLAPP D W, et al. Plexiform neurofibroma genesis: questions of Nf1 gene dose and hyperactive mast cells [J]. Curr Opin Hematol, 2010, 17(4): 287-293. DOI:10.1097/moh.0b013e328339511b.
[8] STASER K, YANG F C, CLAPP D W, et al. Mast cells and the neurofibroma microenvironment[J]. Blood, 2010, 116(2): 157-164. DOI: 10.1182/blood-2009-09-242875.
[9] TRACY T, RICCARDI V M,SUTCLIFFE M, et al. Different patterns of mast cells distinguish diffuse from encapsulated neurofibromas in patients with neurofibromatosis 1 [J]. J Histochem Cytochem, 2011, 59(6): 584-590. DOI: 10.1369/0022155411407340.
[10] YOSHIDA Y, ADACHI K, AYAMAMOTO O. Local mast cell histamine and plasma histamine levels in neurofibromatosis type 1 [J]. Acta Derm Venereol, 2010, 90(6): 637-639. DOI: 10.2340/00015555-0938.
[12] DVORAK A M, MCLEOD R S, ONDERDONK A, et al. Ultrastructural evidence for piecemeal and anaphylactic degranulation of human gut mucosal mast cells in vivo [J]. Int Arch Allergy Immunol, 1992, 99(1): 74-83. DOI: 10.1159/000236338.
[13] BARATELLI F, LE M, GERSHMAN G B, et al. Do mast cells play a pathogenetic role in neurofibromatosis type 1 and ulcerative colitis? [J]. Exp Mol Pathol, 2014, 96(2): 230-234. DOI: 10.1016/j.yexmp.2014.02.006.
[14] ADAMS W, MITCHELL L, CANDELARIA-SANTIAGO R, et al. Concurrent ulcerative colitis and neurofibromatosis type 1: the question of a common pathway [J]. Pediatrics, 2016, 137(2): e20150973. DOI: 0.1542/peds.2015-0973.
[15] TAVAKKOLI H, ASADI M, MAHZOUNI P, et al. Ulcerative colitis and neurofibromatosis type 1 with bilateral psoas muscle neurofibromas [J]. J Res Med Sci, 2009, 14(4): 261-265.
[16] FUKUNAGA S, TAKEDATSU H, MITSUYAMA K, et al. A rare case of ulcerative colitis with neurofibromatosis type 1 [J]. Kurume Med J, 2017, 64(1.2): 25-27. DOI: 0.2739/kurumemedj.ms00014.
[17] AGAIMY A, VASSOS N, CRONER R S. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): clinicopathological spectrum with pathogenetic considerations [J]. Int J Clin Exp Pathol, 2012, 5(9): 852-862.
[18] MIETTINEN M, FETSCH J F, SOBIN L H, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases [J]. Am J Surg Pathol, 2006, 30(1): 90-96.
[19] THWAY K, FISHER C. Diffuse ganglioneuromatosis in small intestine associated with neurofibromatosis type 1 [J]. Ann Diagn Pathol, 2009, 13(1): 50-54. DOI: 10.1016/j.anndiagpath.2007.06.001.
[20] SHEKITKA K M, SOBIN L H. Ganglioneuromas of the gastrointestinal tract. Relation to Von Recklinghausen disease and other multiple tumor syndromes [J]. Am J Surg Pathol, 1994, 18(3): 250-257.
[21] AGAIMY A, SCHAEFER I M, KOTZINA L, et al. Juvenile-like (inflammatory/hyperplastic) mucosal polyps of the gastrointestinal tract in neurofibromatosis type 1 [J]. Histopathology, 2014, 64(6): 777-786. DOI: 10.1111/his.12325.
[22] GOTO K, HIROSAKI T, MASUBUCHI M. Neurofibromatosis type 1-associated inflammatory polyp of the gastrointestinal tract: clinicopathologic analysis of a surgically resected case [J]. Int J Surg Pathol, 2017, 25(1): 65-68. DOI: 10.1177/1066896916648772.
[23] CHAWLA L S, CHINNA J S, DILAWARI JB, et al. Course and prognosis of ulcerative colitis [J]. J Indian Med Assoc, 1990, 88(6): 159-160.
[24] BOCKUS H L, ROTH J L, BUCHMAN E, et al. Life history of nonspecific ulcerative colitis: relation of prognosis to anatomical and clinical varieties [J]. Gastroenterologia, 1956, 86(5): 549-581. DOI: 10.1159/000200623.
[25] POLITIS D S, KATSANOS K H, TSIANOS E V, et al. Pseudopolyps in inflammatory bowel diseases: Have we learned enough? [J]. World J Gastroenterol, 2017, 23(9): 1541-1551. DOI: 10.3748/wjg.v23.i9.1541.
[26] GOLDGRABER M B. Pseudopolyps in ulcerative colitis [J]. Dis Colon Rectum, 1965, 8(5): 355-363.
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
保健与生活(2021年11期)2021-06-10
中国民间疗法(2020年22期)2021-01-07
中国生殖健康(2019年3期)2019-02-01
中华临床免疫和变态反应杂志(2018年6期)2019-01-17
中国当代医药(2015年31期)2015-03-01
中国卫生标准管理(2015年3期)2015-01-27
西南军医(2015年2期)2015-01-22
中华皮肤科杂志(2014年3期)2014-12-19
中国免疫学杂志(2014年5期)2014-02-05
