
N-乙酰半胱氨酸对氧化应激所致胰岛素原成熟障碍的改善作用
2018-05-28 01:06李一卉孙璐戴程婷袁庆新南京医科大学第一附属医院南京210029
山东医药 2018年14期
李一卉,孙璐,戴程婷,袁庆新(南京医科大学第一附属医院,南京210029)
糖尿病是一类由遗传、环境、免疫功能紊乱等因素诱发的严重危害健康的慢性疾病,其病因和发病机制尚未完全明确[1]。研究发现,氧化应激(OS)在糖尿病发病机制中发挥了重要作用[2]。内质网是所有分泌型蛋白和膜蛋白折叠、成熟、储存和转运的场所。近年研究发现,内质网应激(ERS)与糖尿病胰岛β细胞功能减退和胰岛素抵抗密切相关,OS与ERS的相互作用将会加重胰岛β细胞的损伤[3~5]。胰岛素原是胰岛素的前体物质,可裂解为一分子胰岛素和一分子C肽[6]。胰岛素原能否在内质网中正确折叠、进而成熟为构象未成熟胰岛素原复合物(CIPC),对于胰岛素的合成及释放起重要作用。2016年2月~2017年2月,我们观察了抗氧化剂N-乙酰半胱氨酸(NAC)对OS所致胰岛素原成熟及胰岛β细胞分泌胰岛素功能的影响,探讨减轻OS对改善胰岛β细胞功能的作用及机制,为预防糖尿病的发生和发展提供新方向。
1 材料与方法
1.1 细胞与材料 小鼠胰岛细胞系MIN6细胞来源于南京医科大学生物化学系德伟教授实验室。高糖DMEM完全培养基(含有15 %胎牛血清、50 μmol/L β-巯基乙醇、100 U/mL青霉素、100 μg/mL链霉素)、胎牛血清、双抗(Gibco公司,美国);β-巯基乙醇、台盼蓝、过氧化氢(H2O2,Sigma公司,美国);2′7′-二氯荧光素-乙酰乙酸酯(H2DCFDA,Invitrogen公司,美国);胰岛素原单克隆抗体、胰岛素多克隆抗体、管蛋白(Tubulin)多克隆抗体(Abcam公司,英国);胰岛素放免试剂盒(北京北方生物科技研究所,中国)。
1.2 细胞培养 MIN6细胞在高糖DMEM完全培养基中培养,细胞贴壁生长,放入37 ℃、5% CO2培养箱中,每天换液。待细胞密度达75%以上时,按1∶2传代。
1.3 H2O2最佳干预剂量的确定 培养MIN6细胞,生长到对数生长期时,胰酶消化,PBS清洗;终止消化后,加培养基吹匀,然后取等量细胞液种植在6孔板中;待细胞密度达75%左右时,选取不同浓度(0、11 nmol/L、22 nmol/L、44 nmol/L、88 nmol/L及10 μmol/L)H2O2加入培养基,测定不同H2O2浓度时细胞死亡率及蛋白中CIPC含量;发现当H2O2浓度为10 μmol/L时,CIPC含量明显增加,且细胞保持较低的死亡率,从而确定10 μmol/L H2O2作为OS干预的最佳剂量。
1.4 细胞分组及干预方法 分为对照组、H2O2处理组、H2O2+NAC处理组。对照组在培养基中不加入H2O2及NAC处理;H2O2处理组在培养基中加入10 μmol/L H2O2处理;H2O2+NAC处理组培养基中加入10 μmol/L H2O2刺激4 h后,再加入250 μmol/L NAC处理。各组细胞再次放入37 ℃、5 % CO2培养箱中,培养24 h。
1.5 细胞死亡率检测 采用台盼蓝染色法。各组细胞放入37 ℃、5% CO2培养箱中,培养24 h。向各组培养皿中加入台盼蓝试剂,于显微镜下观察细胞生存状态、形态;并对死亡及生存细胞进行手动计数,计算细胞死亡率。
1.6 细胞中活性氧簇(ROS)表达水平检测 采用H2DCFDA荧光探针法。取各组细胞,胰蛋白酶消化,收集细胞;用PBS洗涤后,加入H2DCFDA,放在37 ℃孵育30 min。PBS洗涤3次,荧光酶标仪检测荧光强度,荧光强度越强代表ROS表达水平越高。
1.7 MIN6细胞胰岛素原、CIPC蛋白表达检测 采用Western blotting法。取各组细胞,提取细胞总蛋白;运用非还原Tris-tricine-urea-SDS-PAGE胶进行电泳[7],采用Western blotting法检测细胞中胰岛素原、CIPC蛋白表达。以Tubulin作为内参。计算胰岛素原/CIPC比例。
1.8 细胞上清胰岛素相对含量检测 采用放免法(RIA)。取各组细胞上清,4 ℃ 700 r/min离心10 min,取离心后上清150 μL。采用RIA法检测胰岛素,所用仪器型号为DFM-96型16管手动放射免疫γ计数器,计算各组细胞上清胰岛素相对含量。
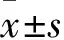
2 结果
2.1 各组细胞死亡率比较 H2O2处理组细胞死亡率高于对照组,H2O2+NAC处理组细胞死亡率低于H2O2处理组(P均<0.05)。见表1。
2.2 各组细胞ROS表达水平比较 H2O2处理组细胞ROS表达水平高于对照组,H2O2+NAC处理组细胞ROS表达水平低于H2O2处理组(P均<0.05)。见表1。
2.3 各组细胞胰岛素原/CIPC比较 H2O2处理组细胞胰岛素原/CIPC低于对照组,H2O2+NAC处理组细胞胰岛素原/CIPC高于H2O2处理组(P均<0.05)。见表1、图1。
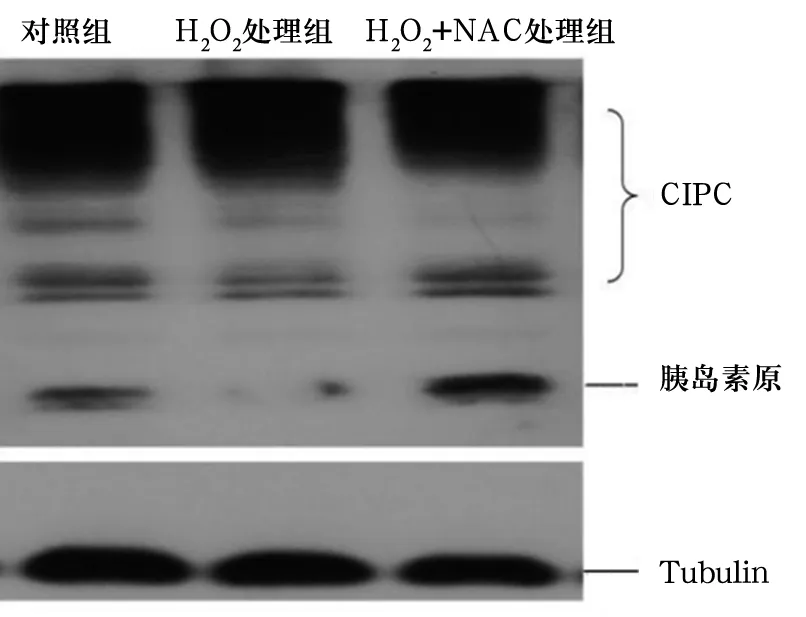
图1 各组细胞胰岛素原、CIPC表达情况(Western blotting法)
2.4 各组细胞上清胰岛素相对含量比较 H2O2处理组细胞上清胰岛素相对含量低于对照组,H2O2+NAC处理组细胞上清胰岛素相对含量高于H2O2处理组(P均<0.05)。见表1。
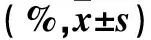
表1 各组细胞检测指标比较
注:与对照组比较,*P<0.05;与H2O2处理组比较,△P<0.05。
3 讨论
糖尿病临床诊断明确时,胰岛β细胞的功能已经损伤明显。如何在糖尿病早期进行有效的干预,是保护胰岛β细胞功能、延缓和预防胰岛β细胞凋亡的重要方法。胰岛素的正确合成是维持血胰岛素水平的基础,首先是编码胰岛素的基因经过转录、翻译,形成前胰岛素原;然后前胰岛素原在内质网剪切信号肽,形成胰岛素原;胰岛素原再移位到高尔基体,剪切成胰岛素和C肽储存在细胞质的囊泡中,最后释放入血[8,9]。近来有学者认为,胰岛素原向胰岛素的转换异常,即正确折叠的胰岛素原减少、CIPC大量堆积,是糖尿病发生的重要原因[10]。
胰岛β细胞具有高度发达的内质网,这与其需要合成和分泌大量的胰岛素及多种糖蛋白相适应,也容易受到各种应激的损伤。其中,OS作为糖尿病发生、发展的重要原因,已被广大学者认可[2]。由于体内氧化与抗氧化作用失衡,从而导致中性粒细胞炎性浸润、蛋白酶分泌增加,产生大量氧化中间产物,这是ROS在体内产生的一种负面作用[11]。内质网的氧化环境对于蛋白质的合成及分泌亦至关重要[12],它参与二硫键的形成、蛋白质的折叠和装配、糖基化、聚糖作用,还是Ca2+贮存的重要场所[6],分泌蛋白的正确折叠对是维持其功能正常的首要条件。未折叠或错误折叠的蛋白不能正常分泌,在细胞堆积时,即通过泛素/蛋白酶体系统、溶酶体系统或通过自噬的方式被降解、清除。胰岛素原在内质网进行合成的过程中,若发生错误折叠,可导致CIPC的产生。一方面,CIPC不能进行下一步剪切,从而使胰岛素合成减少,引起血糖升高。另一方面,胰岛素原因转换为CIPC无法输出而堆积,不仅会引起ERS,还可能使内质网腔的Ca2+渗出;细胞质Ca2+浓度的升高刺激线粒体产生更多的活性ROS,诱发或加重OS[6],OS与ERS的相互作用、恶性循环可能加重β细胞的损伤[5]。所以,蛋白质错误折叠和ROS的产生是紧密相关的事件,但联系二者的机制尚不明确。胰岛素原错误折叠及成熟障碍很可能是OS及ERS引起糖尿病发生、发展的重要原因[13,14]。本研究发现,H2O2处理组与对照组相比,细胞死亡率升高、ROS表达水平升高、胰岛素原/CIPC降低、上清胰岛素含量相对减少。这表明OS加重了胰岛素原的错误折叠并堆积,并可导致胰岛β细胞出现OS损伤、死亡及胰岛素分泌功能障碍。因此,如何减轻OS损伤,从而减少胰岛素原的错误折叠、促进胰岛素原的成熟,可能是保护胰岛β细胞功能、预防糖尿病进展的有效方法。
研究发现,抗氧化剂可以减轻糖尿病症状及血糖水平[15,16]。NAC是含巯基的抗氧化剂,是体内合成谷胱甘肽的原料,能清除体内的ROS,保护许多蛋白质和酶等分子中的巯基不被氧化,但其降糖机制尚不明确。本研究显示,H2O2+NAC处理组较H2O2处理组细胞死亡率降低、ROS表达水平降低、胰岛素原/CIPC升高、上清胰岛素相对含量增加。这提示NAC可以改善H2O2导致的胰岛β细胞损伤,还可以改善OS引起的胰岛素原成熟障碍及胰岛素的释放异常,有效地保护胰岛β细胞功能。
综上所述,NAC可以改善H2O2导致的胰岛β细胞损伤,改善OS引起的胰岛素原成熟障碍及胰岛素的释放异常,有效保护胰岛β细胞功能。因此,胰岛素原成熟障碍、降解增多及相关的ERS和OS可能是糖尿病早期干预的重要靶点。但是,ERS是泛在的保护机制,ERS和OS关系究竟如何,以及抗氧化剂改变OS的具体机制尚待深入研究。
:
[1] Imam K. Clinical features, diagnostic criteria and pathogenesis of diabetes mellitus[J]. Adv Exp Med Biol, 2012,771:340-355.
[2] Simmons RA. Developmental origins of diabetes: the role of oxidative stress[J]. Best Pract Res Clin Endocrinol Metab, 2012,26(5): 701-708.
[3] Brozzi F, Eizirik DL. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes[J]. Ups J Med Sci, 2016,121(2):133-139.
[4] Engin F. ER stress and development of type 1 diabetes[J]. J Investig Med, 2016,64(1):2-6.
[5] Kaneto H. Oxidative stress and ER stress in diabetes[J]. Nihon Rinsho, 2011,69(Suppl 1):171-175.
[6] Petersen OH, Courjaret R, Machaca K. Ca2+tunnelling through the ER lumen as a mechanism for delivering Ca2+entering via store-operated Ca2+channels to specific target sites[J]. J Physiol, 2017,595(10):2999-3014.
[7] Liu M, Hodish I, Rhodes CJ, et al. Proinsulin maturation, misfolding, and proteotoxicity[J]. Proc Natl Acad Sci USA, 2007,104(40):15841-15846.
[8] Kahn SE, Zraika S, Utzschneider KM, et al. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality[J]. Diabetologia, 2009,52(6):1003-1012.
[9] Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death[J]. Science, 2005,307(5708):380-384.
[10] Sun J, Cui J, He Q, et al. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes[J]. Mol Aspects Med, 2015,42:105-118.
[11] Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress[J]. Curr Biol, 2014,24(10):453-462.
[12] Sovolyova N, Healy S, Samali A, et al. Stressed to death - mechanisms of ER stress-induced cell death[J]. Biol Chem, 2014,395(1):1-13.
[13] Yuan Q, Tang W, Zhang X, et al. Proinsulin atypical maturation and disposal induces extensive defects in mouse Ins2+/Akita beta-cells[J]. PLoS One, 2012,7(4):e35098.
[14] Weiss MA. Diabetes mellitus due to the toxic misfolding of proinsulin variants[J]. FEBS Lett, 2013,587(13):1942-1950.
[15] Evans M, Anderson RA, Smith JC, et al. Effects of insulin lispro and chronic vitamin C therapy on postprandial lipaemia, oxidative stress and endothelial function in patients with type 2 diabetes mellitus[J]. Eur J Clin Invest, 2003,33(3):231-238.
[16] Falach-Malik A, Rozenfeld H, Chetboun M, et al. N-Acetyl-L-Cysteine inhibits the development of glucose intolerance and hepatic steatosis in diabetes-prone mice[J]. Am J Transl Res, 2016,8(9):3744-3756.
猜你喜欢
医学信息(2022年9期)2022-11-27
中老年保健(2021年4期)2021-08-22
今日农业(2021年5期)2021-05-22
现代临床医学(2021年1期)2021-01-26
科学之谜(2020年6期)2020-08-11
当代水产(2019年8期)2019-10-12
安徽医科大学学报(2016年12期)2017-01-15
中国病理生理杂志(2015年8期)2015-12-21
——疾病防治的新靶标
中国药理学通报(2015年3期)2015-06-09
中国当代医药(2015年33期)2015-03-01
