
下调大鼠血管内皮细胞血管紧张素转换酶的实验研究
2018-05-11 03:30,,,
中西医结合心脑血管病杂志 2018年7期
,,,
内皮细胞在维持血管内稳态中发挥核心作用,内皮功能障碍将会导致血管收缩、白细胞黏附、胶原蛋白受损、血小板激活、血栓形成、氧化应激及血管壁的炎症反应参与一系列心血管疾病的发生发展[1]。因此,改善内皮功能成为心血管疾病新的治疗靶点。其中肾素-血管紧张素(RAS)系统,尤其是血管紧张素Ⅱ在内皮功能障碍的发生、发展及动脉粥样中发挥核心作用,限制血管紧张素Ⅱ生物活性仍然是心血管疾病治疗的基石[2]。因此,限制调控血管紧张素Ⅱ生成的限速酶血管紧张素转化酶(ACE)成为改善内皮功能的关键。目前,血管紧张素转换酶抑制剂(angiotensin converting enzyme inhibitor,ACEI)以及血管紧张素Ⅱ受体拮抗剂(angiotensinⅡ receptor blocker,ARB)已经被大量临床研究证实能够逆转心血管重构、降低病死率、改善预后,但传统的ACE阻断非血管特异、组织特异、血管壁特异[3]。因此,本研究尝试构建腺病毒载体使其携带有ACE基因短发夹RNA(shRNA),然后通过下调原代培养大鼠血管内皮细胞(ECs)ACE的基因表达,通过ACE基因水平干预RAS系统表达,探索RNAi技术阻断内血管皮细胞ACE的表达在心血管疾病基因治疗领域中的应用前景。
1 材料与方法
1.1 主要材料 已经先期由本研究小组构建完成的p-ACE-sh-RNA质粒,增强的绿色荧光蛋白(EGFP)序列、人源U6启动子,Taq DNA聚合酶、质粒pGenesil-1克隆至两个限制性内切酶的EcoR1和Sac1之间位点。内含有大鼠ACE靶向性的 sh-RNA片段、AdMax腺病毒包装系统主要包括3 Cre、穿梭质粒pDC316-EGFP -shRNA-U6、p BHGlox-E1腺病毒骨架质粒与HEK- 293细胞等(北京本元正阳公司)。T4 DNA连接酶、各种限制性内切酶等。 转染试剂Lipofectamine 2000(Gibco)、RNase A(TaKaRa)、PCR回收纯化产物试剂盒(宁波中鼎公司)。质粒抽提盒(QIAGEN),胎牛血清(Hyclone),氯化铯(Sigma)DMEM,F12培养基(Gibco)。超绝UNIQ-10柱总RNA抽提试剂盒(上海生工生物工程公司)。其余各种试剂均为国产或进口分析纯试剂。探针、引物的设计及合成由上海基康生物技术工程有限公司完成。
1.2 目的基因的获取 采用热休克法将p-ACE-shRNA质粒取1 μL转化入感受态大肠杆菌DH-5α,接种入LB 培养基5 mL中,取出少部分其单克隆,在37 ℃培养振荡约18 h,取3 mL菌液从中提取质粒。在HindIII限制酶和BamHI限制酶共同作用下,在37 ℃水浴酶切中过夜。酶切产物进行1%的琼脂糖凝胶电泳,电泳之后取1 μL 6×Loading Buffer与5 μL酶切产物混匀上样后进行电泳来获取目的基因(10 V/cm)。
1.3 pDC316-EGFP-ACE-U6-shRNA重组质粒的构建 酶切产物进行1%的琼脂糖凝胶电泳,在紫外灯下收集质粒pDC316-EGFP-shRNA-U6的5.2 kb片段酶切产物、质粒pACE-shRNA的60 bp片段酶切产物,回收DNA片段。DNA连接酶在16 ℃水浴连接过夜,用热休克法取连接产物5 μL转化入DH-5α 感受态大肠杆菌,在37 ℃下培养振荡18 h,之后取出菌液3 mL提取质粒,再用酶切等方法筛选出重组正确的质粒,命名为pDC316-EGFP-ACE-U6-shRNA。之后pDC316-EGFP-ACE-U6-shRNA重组质粒再进行酶切鉴定,送上海英骏生物公司进行重组质粒鉴定。
1.4 双质粒共同转染293细胞,同源重组而产生重组腺病毒 在转染的前1 d,293细胞被接种至六孔板中,每孔大约有5×105个细胞,准备DMEM+10% Hyclone的胎牛血清的培养基,置于含有5% CO2的 37 ℃培养箱中培养过夜。待细胞生长到80%~90%底面积时,取穿梭质粒和骨架质粒进行共转染。同时用脂质体Lipofectamine2000进行转染,用培养液DMEM进行稀释,转染之后需要观察出毒迹象。仔细观察后发现出毒现象即为细胞会呈葡萄状,变大变圆,并开始出现明显的噬斑。一定要等待大部分细胞病变而且底部脱落之后再进行收毒。将其命名为Ad5-EGFP-ACE-shRNA。
1.5 将重组腺病毒Ad5-EGFP-ACE-shRNA进行PCR鉴定 293细胞被放在25 cm2的细胞瓶中培养,等到细胞长到底面积90%时,取上述第一代毒种(P1)接种至该细胞上,等到细胞发生完全病变时可按上述方法进行冻融细胞3次,燃后收集已经离心病毒的上清液,即为第二代的毒种(P2)。最后进行目的基因的PCR鉴定,PCR的反应条件为:55 ℃ 30 s,72 ℃ 40 s,94 ℃ 5 min,94 ℃ 30 s,共30个循环;然后72 ℃5 min。之后再进行毒种生产、纯化和滴度测定。
1.6 大鼠ECs的培养与鉴定 首先可以在无菌条件下解剖取出大鼠胸主动脉,然后采用组织贴块法进行血管ECs培养,当细胞长满培养瓶底时即开始传代,免疫荧光Ⅷ因子相关抗原检测阳性即鉴定为ECs,实验采用第3代~第4代的细胞。
1.7 Ad5-EGFP-ACE-shRNA重组腺病毒转染大鼠的血管ECs 当大鼠的血管ECs生长至80%~90%瓶底融合时给予传代,无菌盖玻片的6孔板以5×105个/孔接种放置,细胞同步化后分为空白对照组、阳性病毒组和阴性病毒组3组。在接种后24 h,细胞达60%~80%融合后进行转染,吸去培养板中的培养基,加入等量维持液,选择对数生长期细胞,直接将病毒滴加入培养基中,每孔按照1∶10接种。荧光显微镜下观察48 h后EGFP表达情况。分别在转染前及转染之后24 h、48 h、72 h收集试验细胞。
1.8 实时荧光定量PCR检测ACE mRNA表达 根据参试剂盒说明书来提取总的RNA,然后可以反转录成cDNA。在GenBank上选取大鼠的ACE mRNA序列(序列号NM_012544),ACE探针序列为5’-FTGGCACTTGTCTGTCACTGGAGCCTGATP-3’,上游引物为5’-GCCTCCCAACGAGTTAGAAGAG-3’,下游引物为5’-CGGGACGTGGCCATTATATT-3’;β-actin探针序列为5’-FTCCAGCCTTCCTTCCTGGGTATGGAATC-3’,上游引物为5’-TCAGGTCATCACTATCGGCAAT-3’,下游引物为5’-GGATGTCAACGTCACACTTCATG-3’;探针修饰:P代表TAQMAN-MGB基团,F代表FAM。首先样品的目的基因与管家基因(β-actin)被进行PCR的扩增,之后制备标准的样品曲线。其产物需要进行梯度稀释用于做标准曲线。PCR的反应条件为:94 ℃预变性2 min,94 ℃变性15 s,60 ℃退火、延伸30 s,共30个循环。在GeneAmp 5700 sequence Detector System进行样品扩增同时可以收集荧光信号,之后分析结果。为消除mRNA定量与RT及PCR反应效率的差异对结果的影响,实验中仍然需要同步进行内对照β-actin绝对拷贝数之定量检测,目的用以计算检测基因与β-actin绝对拷贝数之比作为检测基因的相对表达量。

2 结 果
2.1 pACE-shRNA质粒的酶切鉴定(见图1) pACE-shRNA质粒经BamHI和HindIII双酶切预期得到60bp的小片段和4.8kb两条带。结果与预期相符。
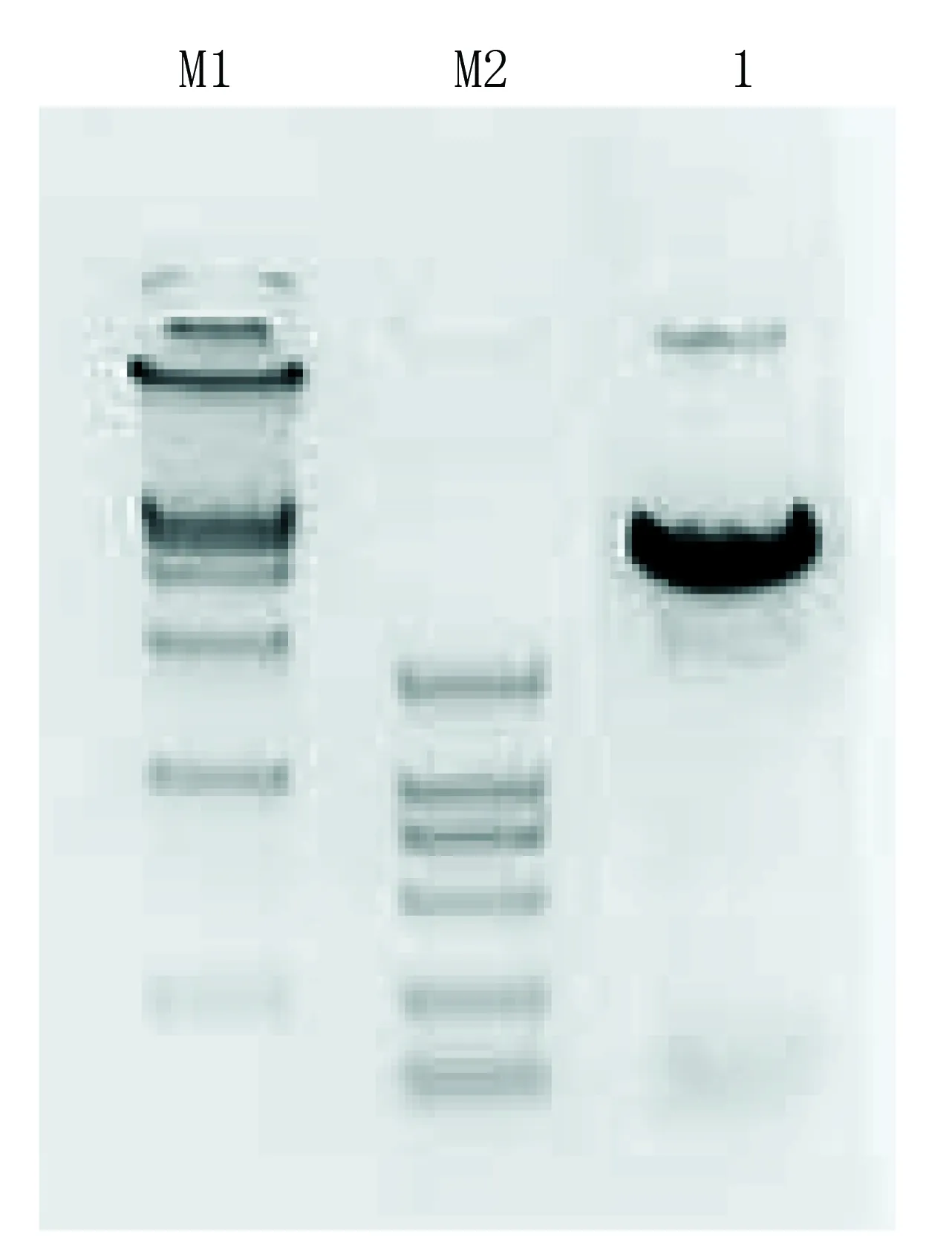
1代表BamHI+HindIII双酶切pACE-shRNA质粒; M1代表DL15 000(上样5 μL,条带大小分别为15000、10000、7500、5000、2500、1000、250);M2代表DL2 000(上样5 μL,条带大小分别为2000、1000、750、500、250、100)。
图1 pACE-shRNA质粒的酶切鉴定电泳图
2.2 酶切鉴定及测序鉴定 pDC316-EGFP-ACE-shRNA-U6重组质粒设计在质粒pDC316-EGFP-ACE-U6-shRNA中shRNA的编码序列之后插入了一个SalI位点,所以,一条线性化条带5.2 kb预计经SalI单酶切产生,得到的电泳结果(见图2)与预计的相符,可以证实该重组质粒经酶切鉴定构建正确。
用引物Cdcksi-r在英骏公司进行测序比对后表明:测出的序列与U6启动子序列完全相同。已经成功将shRNA的序列构建到重组质粒pDC316-EGFP-ACE--U6-shRNA中,因此,重组质粒pDC316-EGFP-ACE-shRNA-U6的构建被证实正确。
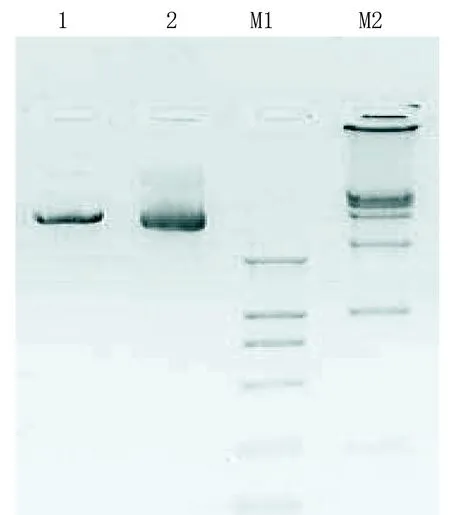
1代表SalI单酶切pDC316-EGFP-ACE-shRNA-U6质粒;2代表pDC316-EGFP-ACE-shRNA-U6质粒;M1代表DL2 000(上样5μL,条带大小分别为2000、1000、750、500、250 、100);M2代表DL15 000(上样5 μL,条带大小分别为15 000、10 000、7 500、5 000、2 500、1 000)。
图2重组质粒pDC316-EGFP-ACE--U6-shRNA酶切鉴定电泳图
2.3 Ad5-EGFP-ACE-shRNA重组腺病毒的PCR的鉴定、测序及滴度测定经鉴定Ad5-EGFP-ACE-shRNA能扩增出目的基因,大小约在500bp左右(见图3)。
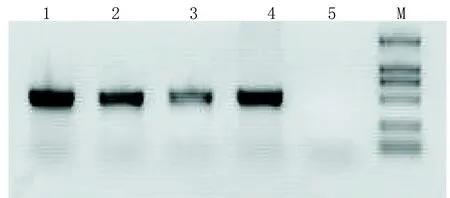
1代表Ad5-EGFP-ACE-shRNA-U6DNA原液;2代表Ad5-EGFP-ACE-shRNA-U6DNA稀释10倍;3代表Ad5-EGFP-ACE-shRNA-U6DNA稀释100倍;4代表阳性对照(pDC316-EGFP-ACE-shRNA-U6);5代表阴性对照;M代表DL2 000Marker(2 000、1 000、750、500、250、100)。
图3 PCR电泳图
DNA测序结果同前比对表明,以位点特异性基因重组方式产生的重组腺病毒所克隆的序列和已知序列完全一致,未发现有移码或突变,表明腺病毒骨架质粒和穿梭质粒发生了重组。
滴度测定结果表明:Ad5对照腺病毒的感染性滴度(TCID50/mL)为7.3×109,Ad5-EGFP-ACE-shRNA 的感染性滴度(TCID50/mL)为 8×109。
2.4 293细胞被Ad5-EGFP-ACE-shRNA重组腺病毒转染结果 293细胞被Ad5-EGFP-ACE-shRNA重组腺病毒转染(见图4),荧光显微镜即可观察到转染48 h后被转染细胞出现绿色的荧光(见图5),表明有GFP的表达。光学显微镜下观察到出现绿色荧光的细胞且细胞内颗粒增多、圆缩、聚集、噬斑形成突起减少,可见“葡萄串”样形态学特征(见图6),有感染能力的腺病毒颗粒被证实包装成功。

图4转染前的293细胞
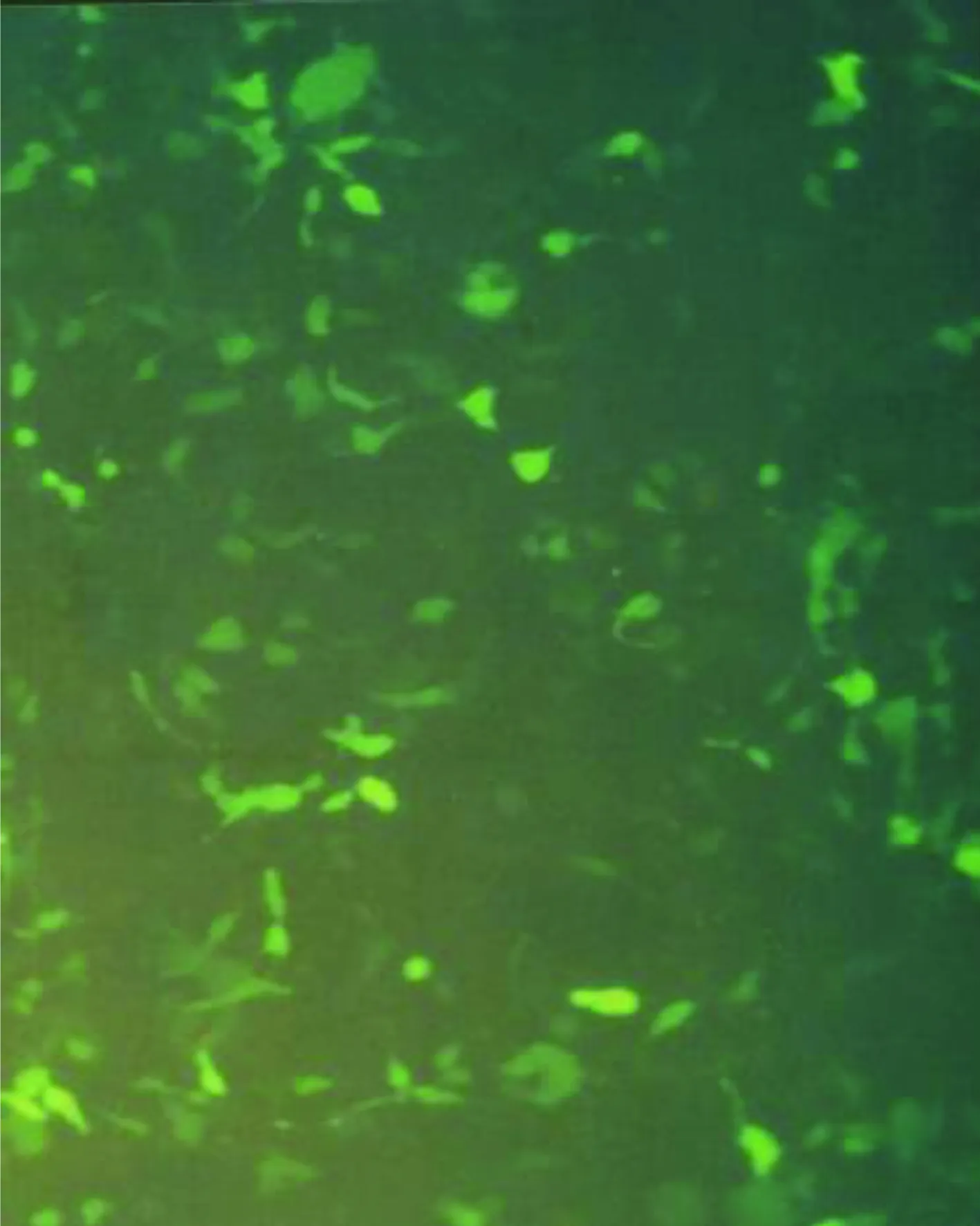
图5重组腺病毒转染293细胞后48 h(荧光显微镜)
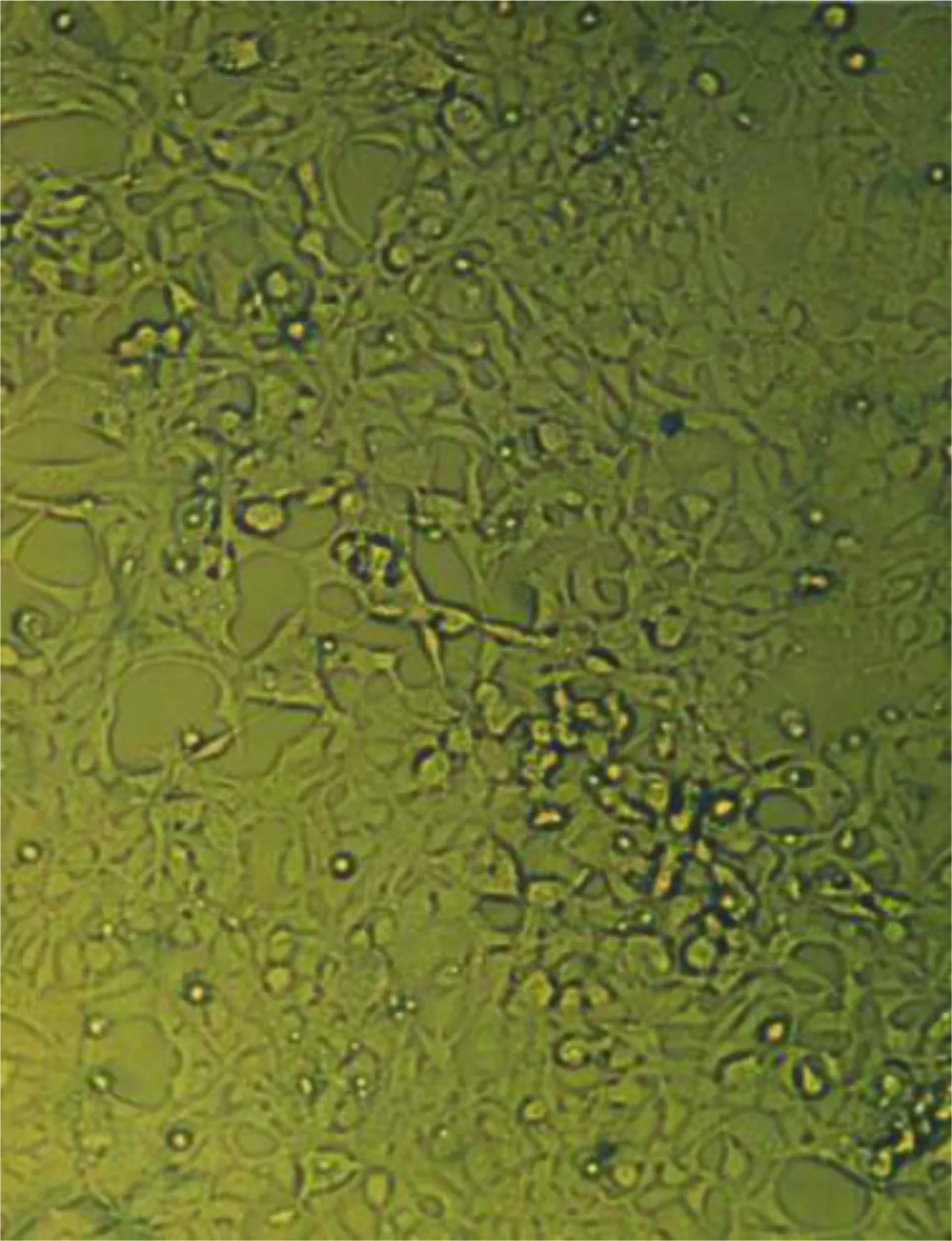
图6 重组腺病毒转染293细胞后噬斑照片(光学显微镜)
2.5 大鼠血管ECs的培养及鉴定 倒置显微镜下观察可见大鼠血管ECs核圆、居中、细胞边界清晰,呈多角形,偶见双核,单层细胞观察到可呈铺路石状排列(见图7)。对原代和传代的细胞进行Ⅷ因子的鉴定:在荧光显微镜下可见细胞呈多边形,胞浆发出绿色荧光,核圆形或椭圆形,边界清晰,大鼠血管ECs被证实培养成功(见图8)。
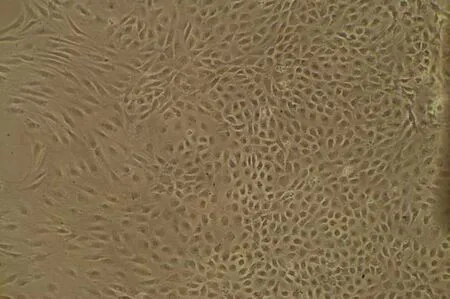
图7 大鼠内皮细胞(×100)

图8大鼠主动脉内皮细胞Ⅷ因子免疫荧光鉴定(×200)
2.6 Ad5-EGFP-ACE-shRNA重组腺病毒转染大鼠血管ECs之后EGFP的表达 Ad5-EGFP-ACE-shRNA重组腺病毒在转染大鼠血管ECs 48 h后即可在荧光显微镜之下观察看到病毒载体所编码的EGFP表达,证实该病毒载体转染细胞成功;同时可见到感染细胞可出现很强的凝聚性并形成细胞团,细胞的折光性增强,有时呈现串状(见图9)。转染效率可达到95%以上。
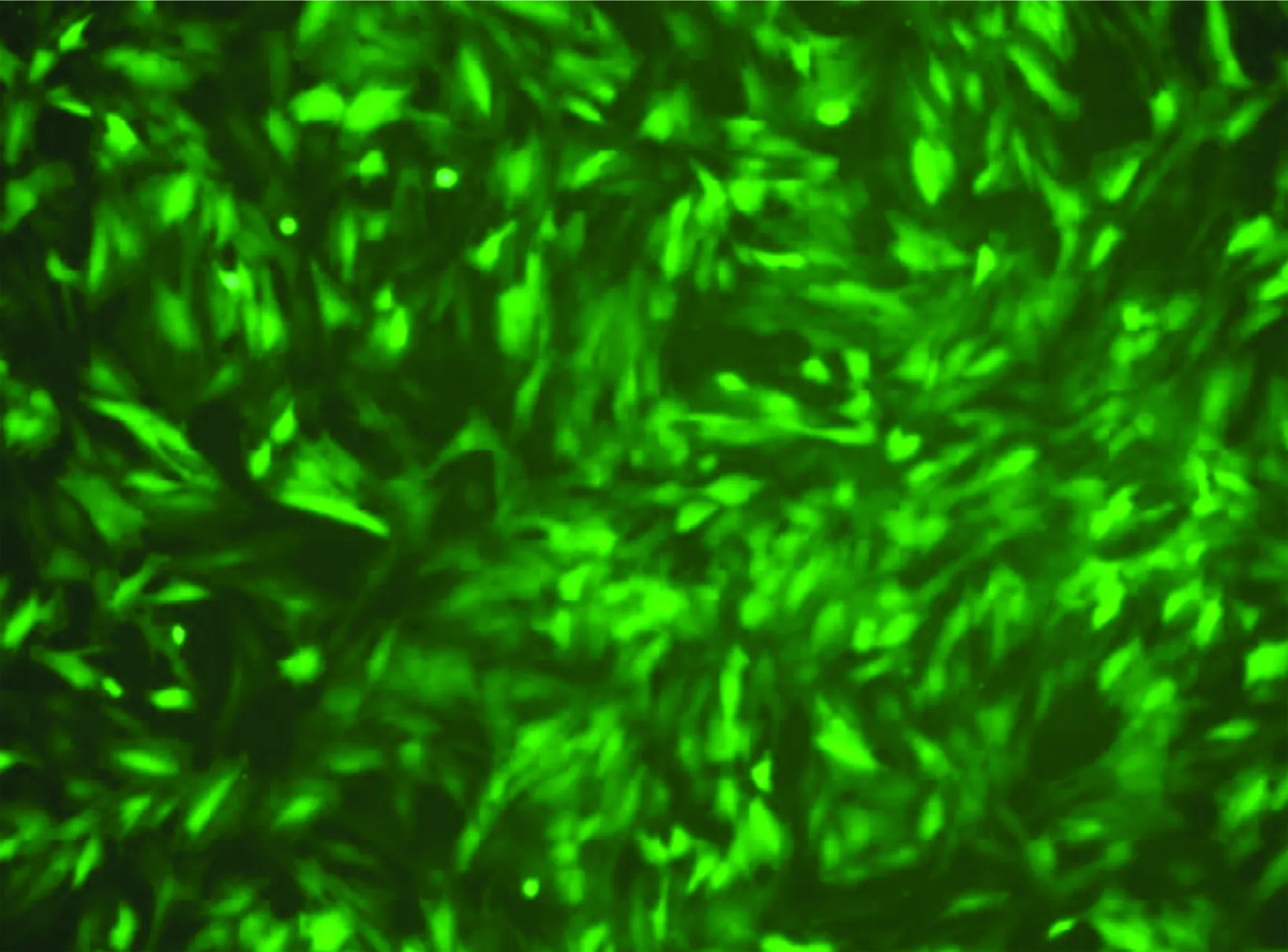
图9 Ad5-EGFP-ACE-shRNA
转染大鼠血管内皮细胞后48 h(×200)
2.7 Ad5-EGFP-ACE-shRNA转染大鼠血管ECs之后对ACE mRNA表达的影响 与空白对照组和阴性病毒组相比,转染之后24 h阳性病毒组Ad5-EGFP-ACE-shRNA细胞的ACE mRNA表达并无明显变化;而48 h显著降低,差异有统计学意义(P<0.01);72 h时更低;表明Ad5-EGFP-ACE-shRNA可以明显下调ACE mRNA的表达。而转染前后各时间点空白对照组和阴性病毒组ACE mRNA表达却无显著变化(见表1)。结果提示RNAi所介导的基因沉默作用主要是通过靶mRNA的下调而产生的。

表1 各组细胞干预前后对ACE/β-actin mRNA表达的影响(±s)
3 讨 论
全世界每年约有1 700万人死于心血管疾病,居死亡原因首位。尽管心血管介入技术及新型药物的研制正在迅猛发展,但目前的治疗现状仍然不是很满意。可喜的是近些年来国际上对RNAi技术的研究已经取得极大进展,通过RNAi技术使致病基因沉默,成功阻断特异性致病基因表达技术已经非常成熟,体内基因敲除技术也正在兴起,该项技术已广泛用于生命科学的多个领域,也为心血管疾病的基因治疗带来希望[4-6]。近些年来 ,国际上多位学者运用RNAi技术对心血管疾病的基因治疗进行了大量研究[7-8]。我们的研究成功地构建了携带有ACE-shRNA基因片段的重组腺病毒载体Ad5-EGFP-ACE-shRNA,并获得了较高的病毒滴度,成功转染了大鼠ECs,有效地抑制了ACE mRNA 表达,证实在原代培养的哺乳动物细胞中RNAi技术能发挥基因下调作用,减少血管紧张素Ⅱ的生成,从而为心血管疾病ACE靶点的基因治疗带来希望,并为心血管疾病基因诊断及治疗带来新的希望。
皮细胞在维持血管稳态中发挥重要作用,是多种心血管疾病发病的重要机制,改善内皮功能对多种心血管疾病均具有极大的保护效应。其中RAS系统,尤其是血管紧张素Ⅱ在内皮功能障碍的发生、发展及动脉粥样中发挥核心作用,限制血管紧张素Ⅱ生物活性无疑是心血管疾病治疗的基石[1-3]。因此限制调控血管紧张素Ⅱ生成的限速酶——血管紧张素转化酶成为改善内皮功能的关键。目前,血管紧张素转换酶以及血管紧张素Ⅱ受体拮抗剂已经被大量临床研究证实能够降低心血管发病率逆转重构、改善预后、降低病死率。
动脉粥样硬化传统的危险因素包括高血压、高脂血症、糖尿病、肥胖、年龄、吸烟、心血管疾病家族史等,已被多位学者公认,传统的危险因素只能解释50%的动脉粥样硬化,有学者发现ECs是动脉粥样硬化的早期表现,它可能是独立于是AS独立的、综合的预测因素。然而近年来研究表明血管紧张素Ⅱ所致内皮细胞功能障碍、血管壁炎症及氧化应激在动脉中发挥重要作用[1,9]。有研究表明,激活内皮细胞表面的血管紧张素Ⅱ会加速NO的降解,减少内皮依赖性血管扩张,从而导致动脉壁损伤,促进内皮细胞表面LDL形成ox-LDL,启动动脉粥样硬化[10]。血管紧张素Ⅱ促进转录因子NF-κB生成,NF-κB通过增加ICAM-1、VCAM-1、E-selectin、MCP-1、IL-1、IL-6生成而促进动脉粥样硬化的发生及进展[11-14];通过刺激NADP(H)和黄嘌呤氧化酶的活性而诱导氧自由基产生,加速动脉粥样硬化进程[15];通过激活诱导动脉粥样硬化斑块纤维帽MMPs,导致纤维帽破裂从而诱发ACS[16];本研究从基因水平阻断ACE表达,减少血管紧张素Ⅱ的生成,理论上可以阻断炎症、氧化应激及基质金属蛋白酶活动在动脉粥样硬化中的作用,有望为冠心病ACE靶点的基因治疗带来希望。
高血压是全世界发病率最高的疾病之一,高血压的发病是综合的多因素疾病,内皮功能障碍与高血压密切相关。其中RAAS系统通过调节内皮细胞功能在高血压发病中发挥极为重要的作用[17]。最近目前已经有不少学者利用miRNA技术研究了RAAS相关的miRNA对高血压的影响。Marques等[18]发现遗传性高血压病人交感神经过度激活,miR-181a 表达降低时,肾素生成增多,血压升高。因此miR-181a 表达水平增加时会通过抑制交感神经系统活性、减少肾素分泌来降低血压; Hu等[19]研究发现miR-145过度表达会可下调ACE蛋白水平表达,导致血压下降,但这种途径并不会引起ACE mRNA的减少。相反激活ERK1/2 通路将会抑制 miR-145表达,上调ACE的表达水平导致血压升高; Eskildsen等[20]体内、体外实验均证明miR-132、miRNA-212水平和血压水平正相关。同时发现miR-132、miR-212,通过激活Gαq依赖的信号转导通路参与AngⅡ介导的高血压的发生;最近的研究表明miR-155表达水平增高可下调ATG1RmRNA使血压下降[21];miR-124、miR-135a能够抑制盐皮质激素受体基因表达减轻前负荷使血压下降[22-23];上述研究均为高血压的基因诊断及治疗带来希望,但是本研究运用RNAi技术降解ACE mRNA,下调ACE,可使血压水平下降。且RNAi技术相比miRNA技术高效、更特异、更持久,周期短、成本低[4-6],降压效果及靶器官保护效应可能比上述研究更持久,目前运用RNAi技术沉默内皮细胞ACE基因的研究报道甚少,本研究有望为高血压的基因治疗提供新的思路。
参考文献:
[1] Matsuzawa Y,Lerman A.Endothelial dysfunction and coronary artery disease:assessment,prognosis and treatment[J].Coronary Artery Disease,2014,25(8):713-724.
[2] Savoia C,Schiffrin EL.Inhibition of the renin angiotensin system:implications for the endothelium[J].Curr Diab Rep,2006,6:274-278.
[3] Cole J,Quach DL,Sundaram K,et al.Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure[J].Circ Res,2002,90:87-92.
[4] Fischer SE.RNA Interference and microRNA-mediated silencing[J].Curr Protoc Mol Biol,2015,112:26.
[5] Li Z,Rana TM.Molecular mechanisms of RNA-triggered gene silencing machineries[J].Accounts of Chemical Research,2012,45(7):1122-1131.
[6] Koenig O,Walker T,Perle N.New aspects of gene-silencing for the treatment of cardiovascular diseases[J].Pharmaceuticals,2013,6:881-914.
[7] Wan W,Jiang X,Li X,et al.Silencing of angiotensin converting enzyme by RNA interference prevents H9c2 cardiomyocytes from apoptosis induced by anoxia/reoxygenation through regulation of the intracellular renin-angiotensin system[J].International Journal of Molecular Medicine, 2013, 32(6):1380.
[8] Zhang L,Erfle H, Harder N,et al.High-throughput RNAi screening identifies a role for the osteopontin pathway in proliferation and migration of human aortic smooth muscle cells[J].Cardiovascular Drugs & Therapy, 2016, 30(3):281-295.
[9] Brunner HR,Gavras H.Angiotensin blockade for hypertension:a promise fullled[J].Lancet,2002,359:990-992.
[10] Limor R,Kaplan M.Angiotensin Ⅱ increases the expression of lectin-like oxidized low-density lipoprotein receptor-1 in human vascular smooth muscle cells via a lipoxygenase-dependent pathway[J].Am J Hypertents,2005,18(3):299-307.
[11] Zhang X,Wu M,Jiang H,et al.Angiotensin Ⅱ upregulates endothelial lipase expression via the NF-Kappa B and MAPK signaling pathways[J].PLoS One,2014,9(9):e107634.
[12] Zhu L,Carretero OA,Xu J,et al.Activation of angiotensin Ⅱ type 2 receptor suppresses TNF-α-induced ICAM-1 via NF-κB:possible role of ACE2[J].American Journal of Physiology - Heart and Circulatory Physiology,2015,309(5):H827-H834.
[13] Pandey A,Goru SK,Kadakol A,et al.Differential regulation of angiotensin converting enzyme 2 and nuclear factor-κB by angiotensin Ⅱ receptor subtypes in type 2 diabetic kidney[J].Biochime,2015,118:71-81.
[14] Deng B,Fang F,Yang T,et al.Ghrelin inhibits AngⅡ-induced expression of TNF-α,IL-8,MCP-1 in human umbilical vein endothelial cells[J].International Journal of Clinical and Experimental Medicine,2015,8(1):579-588.
[15] Dikalov SI,Nazarewicz RR.Angiotensin Ⅱ-induced production of mitochondrial reactive oxygen species:potential mechanisms and relevance for cardiovascular disease[J].Antioxidants & Redox Signaling,2013,19(10):1085-1094.
[16] Yaghooti H,Firoozrai M,Fallah S,et al.Angiotensin Ⅱ differentially induces matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 production and disturbs MMP/TIMP balance[J].Avicenna Journal of Medical Biotechnology,2010,2(2):79-85.
[17] Poulter NR,Prabhakaran D.Renin-angiotensin system and cardiovascular risk[J].Lancet,2007,369:1208-1219.
[18] Marques FZ,Campain AE,Tomaszewski M,et al.Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for micro RNAs[J].Hypertension,2011,58:1093-1098.
[19] Hu B,Song JT,Qu HY,et al.Mechanical stretch suppresses micro RNA-145 expression by activating extracellular signal-regulated kinase 1/2 and upregulating angiotensin-converting enzyme to alter vascular smooth muscle cell phenotype[J].PLoS One,2014,9:e96338.
[20] Eskildsen TV,Jeppesen PL,Schneider M,et al.Angiotensin Ⅱ regulates micro RNA-132/-212 in hypertensive rats and humans[J].Int J Mol Sci,2013,14:11190-11207.
[21] Sethupathy P,Borel C,Gagnebin M,et al.Human micro RNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3' untranslated region:a mechanism for functional single-nucleotide polymorphisms related to phenotypes[J].AmJ Hum Genet,2007,81:405-413.
[22] Luo JQ,Wang LY,He FZ,et al.Effect of NR3C2 genetic polymorphisms on the blood pressure response to enalapril treatment[J].Pharmacogenomics,2014,15:201-208.
[23] Berger S,Bleich M,Schmid W,et al.Mineralocorticoid receptor knockout mice:pathophysiology of Na+metabolism[J].Proc Natl Acad Sci U S A,1998,95:9424-9429.
猜你喜欢
体育科技文献通报(2022年4期)2022-10-21
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
世界科学技术-中医药现代化(2022年3期)2022-08-22
成都医学院学报(2022年4期)2022-08-19
传染病信息(2022年3期)2022-07-15
中国动物保健(2022年2期)2022-05-05
养殖与饲料(2021年11期)2021-11-15
现代临床医学(2021年5期)2021-11-02
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
