
铑靶康普顿散射校正-X射线荧光光谱(XRF)法测定铝用炭素阳极材料中的微量元素
2018-04-11 11:04:05白万里马慧侠
中国无机分析化学 2018年2期
白万里 马慧侠 刘 静 彭 展
(中国铝业 郑州有色金属研究院有限公司,郑州 450041)
前言
铝用炭素材料是指以煅后石油焦、沥青焦或无烟煤等为主要骨料,以煤沥青等作黏结剂制成的糊类或块类炭素制品,主要作为金属铝电解生产过程中电解槽的阳极和阴极。由于阴极材料不属于大量消耗性材料,因此铝用炭素材料微量元素的测定主要指铝用炭素阳极材料,主要包括石油焦、煅后焦、预焙阳极等。铝用炭素阳极材料用量巨大,在我国每年铝用炭素材料生产中石油焦使用量超过2 000万t,预焙阳极生产量也已经超过1 500万t。
铝用炭素阳极材料中微量元素测定主要包含硫、钒、钠、钙、硅、铁、镍等元素,各类原料中元素含量范围相对较稳定。铝用炭素材料中微量元素的测定主要有分光光度法[1]、电感耦合等离子体原子发射光谱(ICP-AES)法[2]和X射线荧光光谱(XRF)法[3-4]。但分光光度法和ICP-AES法中样品需要经过高温灰化、熔融、溶解等繁琐步骤,样品转移次数较多,极易造成误差,所以该方法应用不大;XRF法测定铝用炭素材料中微量元素含量,制样极其简便,只需在炭素样品中加入合适的黏结剂研磨、压片即可,并且可实现多元素的同时测定分析,因此XRF法是当今测定铝用炭素微量元素最主要的方法。通过对比各种常用黏结剂中杂质元素含量,选择硬脂酸作混合黏结剂,用X射线荧光光谱法测定铝用炭素阳极材料中微量元素硫、钒、钠、钙、硅、铁、镍、钛、铝、镁、磷、铅、锌、铬、锰含量;优化铅元素的测定条件,采用经验系数法和铑靶康普顿散射内标校正法相结合来校正工作曲线,数据表明铁、镍、铅、锌、铬、锰等重金属元素铑靶康普顿散射法效果优于经验系数法[5-7];未知样品的检测结果与标准值测定结果相吻合,准确度和精密度实验表明该方法可靠性和重现性良好。
1 实验部分
1.1 仪器与试剂
XRF 1800波长色散X射线荧光光谱仪(日本岛津公司);ZM-1振动磨(配碳化钨研磨料钵,中国科学院长春光学精密机械与物理研究所);YYJ-40半自动压片机(中国科学院长春光学精密机械与物理研究所);BP210电子分析天平,感量为0.1 mg(北京赛多利斯公司)。
硬脂酸(分析纯);硼酸(优级纯);甲基纤维素(分析纯);淀粉(分析纯)。
各元素测定条件见表1。

表1 元素测定条件Table 1 Measurement condition for elements
1.2 实验方法
称取12 g试样(精确到0.1 mg),加入2 g(精确到0.1 mg)硬脂酸作黏结剂,于振动磨上振磨20 s,然后样品全部放入压片机,在196 kN压力下静态保压20 s制样,卸压后取出样片,用洗耳球吹去表面颗粒后待测。
1.3 校准样品
选用瑞士R&D炭素公司的生焦系列C311-C355、煅后焦系列PC101-PC110以及中铝郑州研究院的GPW系列GPW1-GPW9等标准样品,按照上述方法和条件制备标准样品,制作工作曲线对石油焦、煅后焦和预焙阳极等材料进行测定,元素测定范围见表2。
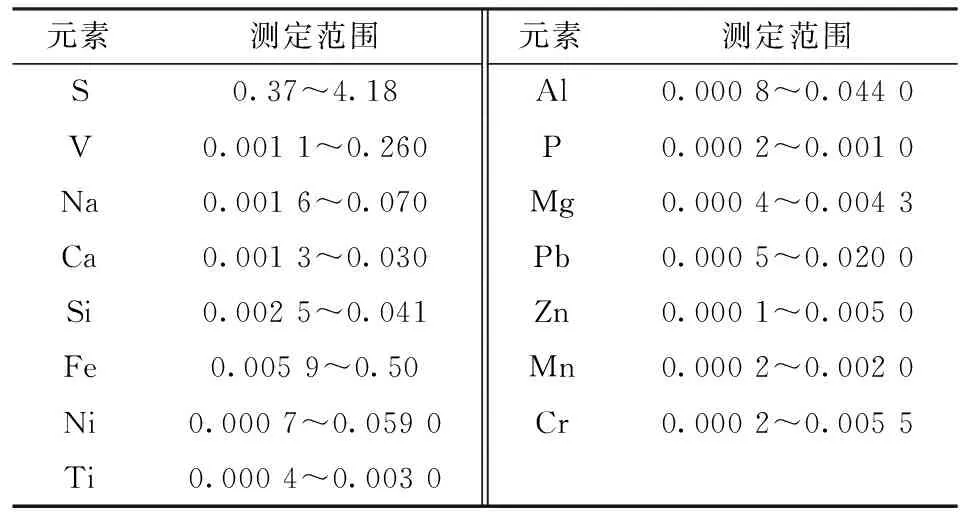
表2 标准样品元素含量范围Table 2 Content range of each element instandard samples /%
2 结果与讨论
2.1 黏结剂种类
压片法是X射线荧光光谱法最常用的制样方法,为确保压片效果良好,研磨中会添加黏结剂,但诸多压片法的国家标准或行业标准中往往一味追求黏结剂的黏结性能,而忽略黏结剂本身所含杂质元素带来的污染或者测定偏差,所以压片法所用的黏结剂除了黏结性良好之外其本身微量元素也必须在合理范围之内。
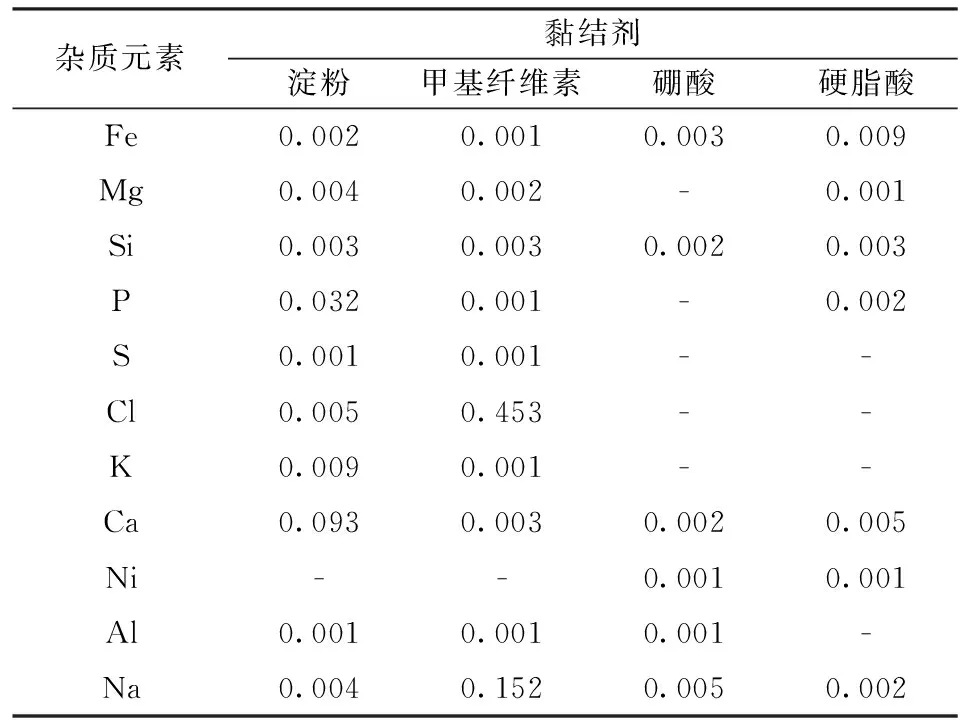
为了防止黏结剂中钠、钙、铁等微量元素的含量影响待测元素结果,先将淀粉、甲基纤维素、硼酸、硬脂酸直接在压片机上压片,用X射线荧光光谱仪定性-半定量程序测定其微量元素的含量,压片压力、保压时间以及测定条件均相同,结果见表3。
经过测量发现淀粉中钙和磷的含量较高、甲基纤维素中钠含量较高,会污染样品,同时淀粉和甲基纤维素极易吸潮,测定中出现真空度下降,测定中断现象;硬脂酸中铁含量约为0.009%,但是硬脂酸添加量较少,按照6∶1比例添加黏结剂后引进的铁元素只有0.001%级别;硼酸微量元素含量极低也可以选取。
表3黏结剂中杂质元素含量
Table3Contentofimpurityelementinbinder/%
为验证各黏结剂的黏结性能,称取4份同一预焙阳极试样各12.0 g,分别加入2.0 g淀粉、硼酸、甲基纤维素、硬脂酸作黏结剂,碳化钨料钵研磨后用压片机压片制样。研磨时间、压片条件均保持一致。甲基纤维素、硼酸样片外观有轻微起伏,淀粉、硬脂酸压片外形良好,但是用手指分别轻轻划动测量面,发现淀粉样片划痕较大更容易掉渣,硬脂酸样片没有掉渣现象划痕较弱,因此从黏结性能来讲,硬脂酸黏结性能较其它黏结剂更强。综合考虑,选用硬脂酸作为黏结剂。
2.2 黏结剂用量
铝用炭素阳极材料中石油焦含有挥发分,相对于阳极和煅后焦最容易黏结和压片,因此如果加入黏结剂研磨后预焙阳极和煅后焦能够很好地压片,石油焦必然也能很好地研磨压片(但应该注意加入量过大造成料钵黏结现象)。
为验证黏结剂用量,称取4份同一预焙阳极试样各12.00 g,分别加入0.50、1.00、2.00、3.00 g硬脂酸作黏结剂,碳化钨料钵研磨后用压片机压片制样。其中0.50 g用量的样片勉强成型,表面有凸起,侧面有裂缝,样片外观不好,不能使用,其余的都能压制成片,样片表面紧密度随着硬脂酸用量加大而加大,但研磨中料钵黏接程度也越来越明显。为验证石油焦样品黏结剂用量,称取4份同一石油焦试样各12.0 g,分别加入0.50、1.00、2.00、3.00 g硬脂酸作黏结剂,样片均能压片成形,表面硬度与致密度也随用量加大而加大,但是3.00 g用量的样片在研磨时黏接特别明显,不易清洗。
综合考虑,制样时12.00 g样品称取2.00 g硬脂酸黏结剂。
2.3 研磨时间
称取4份同一预焙阳极试样各12.00 g,加入2.00 g硬脂酸作黏结剂,加入碳化钨料钵用新设备分别研磨10、20、30、40 s后样品全部放入压片机制样。随着时间增加,在研磨中样片黏结料钵程度逐步增加,压片后样表面的硬度与质密度也逐步增大,同一测定条件下将4个样片放入X射线荧光仪中测定各元素的荧光强度,数据见表4。

表4 研磨时间对计数率的影响Table 4 The effect of milling time oncounting rate /kcps
表4数据表明,研磨时间为10 s的样片中多种元素强度是最小的,研磨时间为20、30 s的荧光强度差别不大,40 s荧光强度最大。但随着研磨时间增长,料钵中进入到样品中的钨、钴元素逐步增多,钨元素过高可能会通过影响锌元素的背景测定最终影响测定结果,ZnKα和WLα谱线见图1。综合各因素,最终选择研磨时间为20 s。

图1 ZnKα和WLα线谱图Figure 1 Spectrogram of ZnKα and WLα.
综上所述,样品的制备方法为:称12 g的试样(精确到0.1 mg),加入2 g(精确到0.1 mg)硬脂酸作黏结剂,于振动磨上振磨20 s,然后样品全部放入压片机,在196 kN压力下静态保压20 s制样,卸压后取出样片,用洗耳球吹去表面颗粒后待测。
2.4 铅元素测定线的选择
X射线荧光光谱法测定铅元素可以选择PbLα或PbLβ1作分析线,通常PbLα计数率绝对强度要高于PbLβ1。因此有文献用PbLα作分析线[8-9]。图2为PbLα和PbLβ1线谱图。

图2 PbLα和PbLβ1线谱图Figure 2 Spectrogram of PbLα and PbLβ1.
选择三个样片测其PbLα、PbLβ1强度,单点扣背景法[10]计算净强度时,PbLα和PbLβ1强度非常接近,甚至部分样品PbLβ1高于PbLα,结果见表5;同时选择LiF200作分光晶体时,PbLα线的2θ数值为34.00,AsKα线的2θ数值为33.93,两者几乎完全重叠,潜在的砷元素会直接影响铅元素的荧光强度。综合荧光强度和谱线重叠双重因素,最终选择PbLβ1作为测定线。
2.5 校准曲线及校正
2.5.1背景选择与校正数学模型
校准曲线是由标准样品中各元素荧光强度R(kcps)与元素含量C(%)的线性回归方程,岛津XRF 1800设备中,软件采用德杨数学模型(de Jongh),其表达式为:
(1)
式(1)中:Ci是元素的浓度(%)或计数率(cps);Di、Ei分别是校准曲线的截距与斜率;Ri是元素的荧光强度(cps),αij为经验影响系数,经验系数只有数学上的统计意义,无明确的物理意义,不能转移使用[11]。
2.5.2元素校正
铝用阳极材料基体为碳元素,属于超轻基体,测定轻基体微量、痕量元素时散射背景则可以作为内标参比通道,对分析有良好的补偿效果,铑靶康普顿散射内标校正法就是其中一种重要的方法。

表5 PbLα与PbLβ1荧光强度Table 5 The fluorescence intensity of PbLα and PbLβ1
铑靶康普顿散射校正法使用是有条件的,首先分析线波长要位于样品主要基体元素吸收线波长的短波侧,其次铑靶相干散射线RhKαC波长与分析线波长越接近,则分析线与内标线搭配越理想,补偿效果也越好[11],重元素分析波长离RhKαC相对与轻元素近,因此康普顿散射内标法主要考虑对重元素进行校正。炭素材料基体为碳元素,其吸收限波长为4.389 nm,TiKα的波长为0.275 nm,VKα的波长为0.251 nm,CrKα的波长为0.229 nm,MnKα的波长为0.210 nm,FeKα的波长为0.194 nm,NiKα的波长为0.166 nm,ZnKα的波长为0.144 nm,PbLβ1的波长为0.098 nm,测定元素的波长均在基体元素吸收限的短波侧,并且与RhKαC波长0.064 nm较接近。通过不加康普顿散射内标校正以及使用内标法对重元素Ti、V、Cr、Mn、Fe、Ni、Zn、Pb进行曲线回归计算,相关参数见表6。

表6 重元素校准曲线系数比较Table 6 Comparison of calibration curvefor heavy element
表6数据中V元素在经过铑靶康普顿散射内标校正后准确度系数和相关系数均下降,Ti元素的准确度系数下降,校正系数略微提高,可能是由于Ti和V元素分析线波长与RhKαC波长相距较远,两者匹配性不好的缘故。最终选择Cr、Mn、Fe、Ni、Zn、Pb进行铑靶康普顿散射内标,其余采用经验系数校正。校正后各元素准确度系数以及相关系数见表7。

表7 校准曲线系数Table 7 Coefficients of the calibration curve
2.6 精密度实验
选取4个炭素样品,分别制取11个样片,将测定结果进行统计,求得各元素测定结果的标准
偏差(SD)和相对标准偏差(RSD),结果见表8。各元素各个梯度的RSD均小于8%,最高的钠元素和钛元素也是在10%左右,证明方法重现性良好。
2.7 准确度实验
以不参与回归曲线的标准样品GPW-6、GPW-8、PC-105、C-353进行准确度实验,结果见表9,由表9数据对照可知本方法测定结果和认定值相一致。
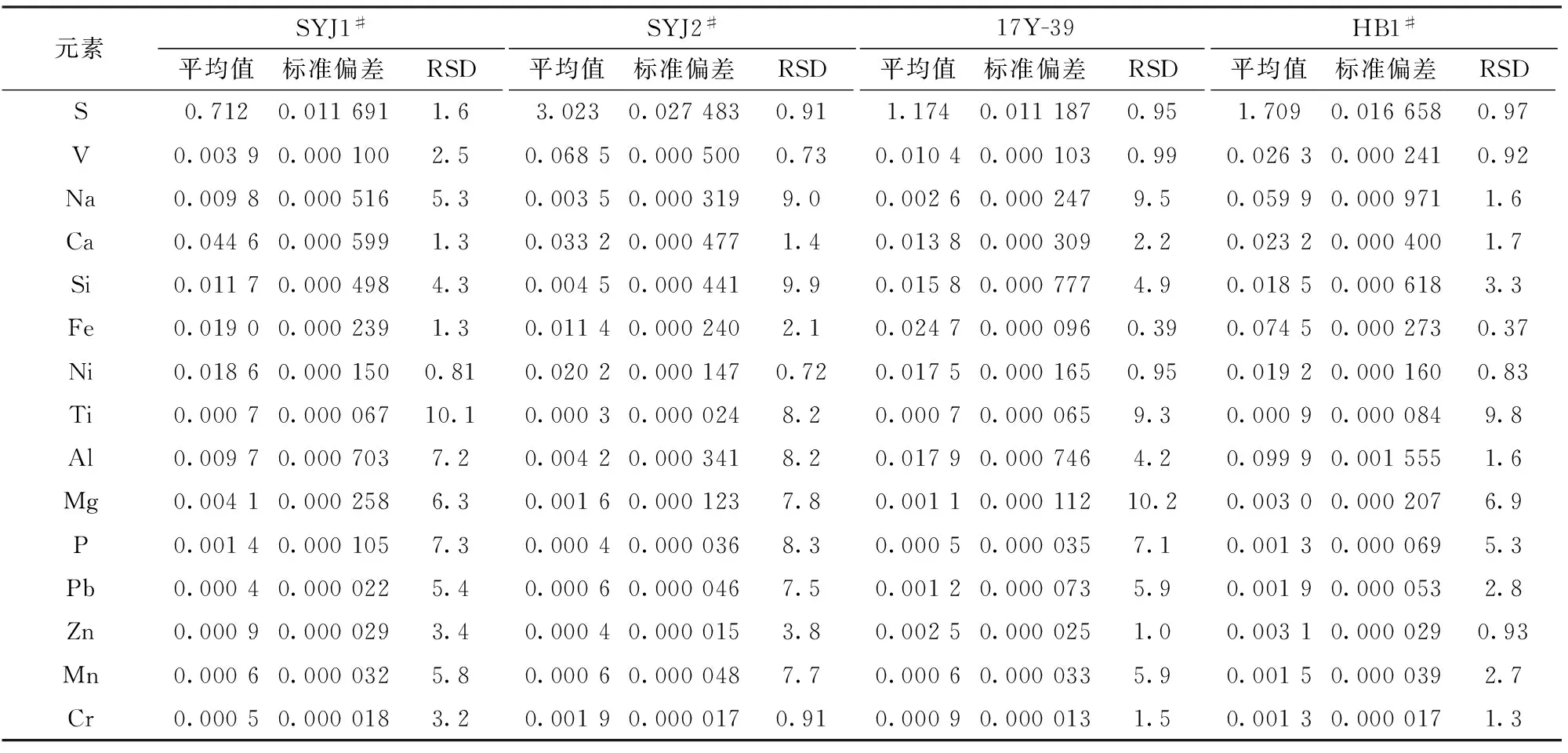
表8 精密度实验结果Table 8 Precision test results /%
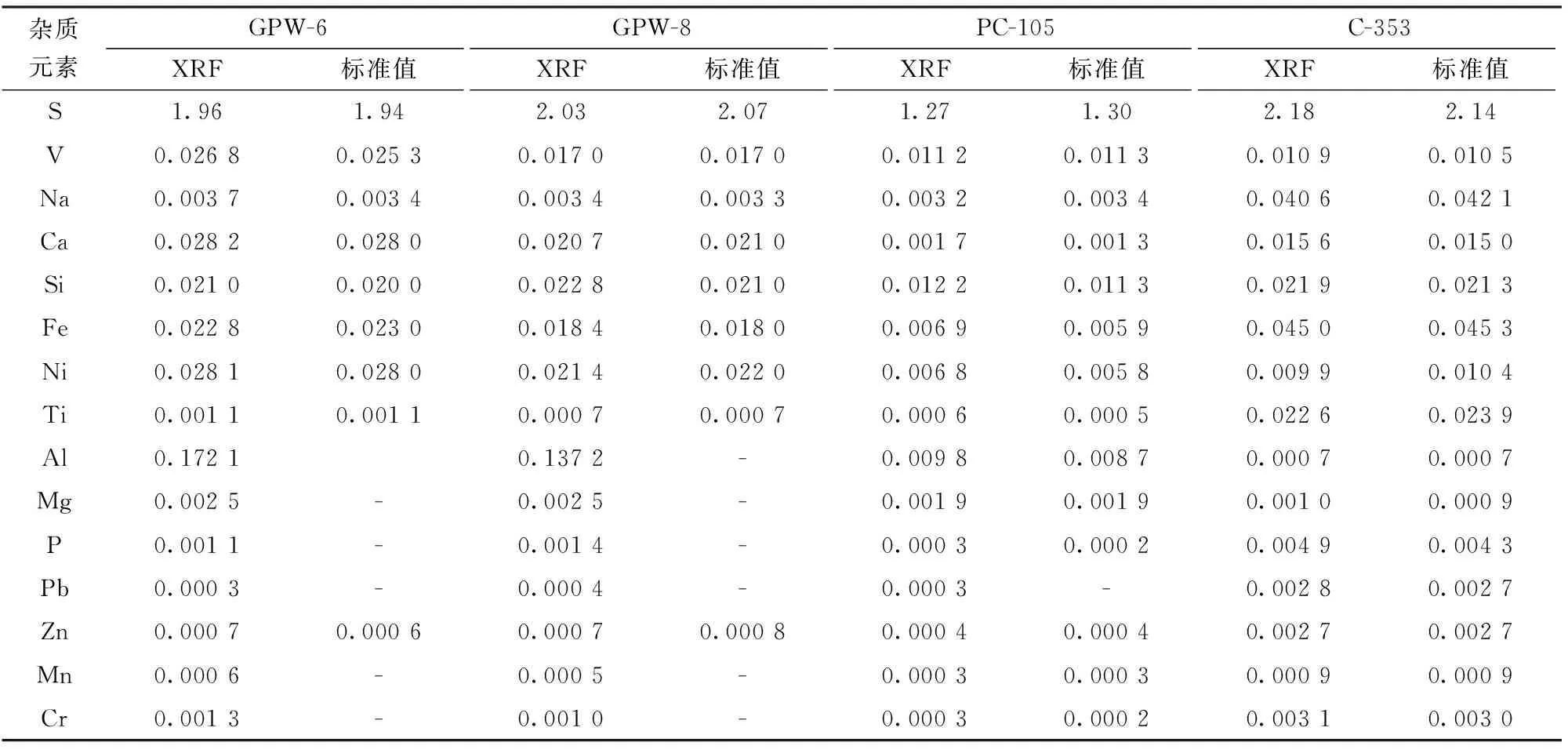
表9 标准样品的准确度实验结果Table 9 Accuracy of test results of the standard sample w/%
3 结论
铝用炭素阳极材料X射线荧光光谱压片法要加入黏结剂,而黏结剂中杂质元素往往是被忽略的一个环节,因此需要筛查黏结剂中杂质元素,以免影响测定结果,即使同一种经常使用的黏结剂也应对不同批次予以杂质元素筛查,这也对所有需要添加黏结剂的压片法有借鉴意义;采用PbLβ1作铅元素分析线可以提高荧光强度,避免砷元素的干扰,提高分析灵敏度;采用经验系数法和铑靶康普顿散射内标校正法相结合来校正工作曲线,重元素的分析精度得到提高。
[1] 中国石油化工总公司.SH/T 0058—1991石油焦中硅、钒和铁含量测定法[S].北京:中国标准出版社,1991.
[2] 中华人民共和国国家发展与改革委员会,中国国家标准化管理委员会.YS/T 587.5—2006炭阳极用煅后石油焦检测方法 第5部分:微量元素的测定[S].北京:中国标准出版社,2006.
[3] 中华人民共和国国家发展与改革委员会,中国国家标准化管理委员会.YS/T 63.16—2006铝用碳素材料检测方法 第16部分:微量元素的测定 X射线荧光光谱分析方法[S].北京:中国标准出版社,2006.
[4] ISO/TC47/SC7. ISO 12980: 2000 Carbonaceous materials in the production of aluminium—Green coke and calcined coke for electrode[S]. First edition.Switzerland ,2000-04-01.
[5] 孙晓飞,文孟喜,杨丹丹.康普顿散射线结合经验系数法校正在X射线荧光光谱测定石灰石和白云石中的应用[J].冶金分析(MetallurgicalAnalysis),2016,36(1):11-17.
[6] 张庆建,岳春雷,孙瑞昌,等. X射线荧光光谱法测定煤中砷磷氯[J].冶金分析(MetallurgicalAnalysis),2015,35(7):84-88.
[7] 严家庆,杨霖,张剑鸣,等. X射线荧光光谱法测定煤中氯[J].煤质技术(CoalQualityTechnology),2009(6):22-23.
[8] 张爱芬,马慧侠,李新华. X射线荧光光谱测定铝用炭素材料中微量元素[J].冶金分析(MetallurgicalAnalysis),2008,28(4):27-30.
[9] 马慧侠,张爱芬,李智慧.预焙阳极微量元素XRF测定方法的研究[J].分析测试学报(JournalofInstrumentalAnalysis),2008,27(9):998-1001.
[10] 罗立强,詹秀春,李国会.X射线荧光光谱仪[M].北京:化学工业出版社,2008:59-60.
[11] 高新华,宋武元,邓赛文,等.实用X射线光谱分析[M].北京:化学工业出版社,2016:215-228.
猜你喜欢
食品工业(2022年6期)2022-07-04 08:39:14
食品工业科技(2022年10期)2022-05-14 08:26:26
一重技术(2021年5期)2022-01-18 05:41:58
有色金属设计(2021年4期)2022-01-09 11:34:44
人大建设(2019年6期)2019-10-08 08:55:58
计算机与数字工程(2019年1期)2019-03-01 02:51:32
宇航计测技术(2018年5期)2019-01-03 02:54:52
农业知识(2018年35期)2018-09-26 09:22:04
纺织学报(2018年9期)2018-09-23 01:26:48
西部皮革(2018年2期)2018-03-05 08:41:46
