
异呋咱类熔铸炸药载体分子设计与性能研究
2018-03-01 01:07杜黎小松刘玉存程桂敏荆苏明罗进王萌霞
兵工学报 2018年1期
杜黎小松, 刘玉存, 程桂敏, 荆苏明, 罗进, 王萌霞
(中北大学 环境与安全工程学院, 山西 太原 030051)
0 引言
火炸药技术是推动现代战略武器和战术武器发展、实现军事装备改革的重要基础技术,火炸药性能的优劣直接决定着武器装备的生存能力和作战时效[1]。在军用混合炸药中,熔铸炸药由于其综合性能优良、制造简单、成本低廉和装配方便等优点而成为各种榴弹、破甲弹、火箭弹等战斗部的主装药,是各国应用最广泛的一类军用混合炸药。在我国的熔铸炸药中,以三硝基甲苯(TNT)和2,4-二硝基苯甲醚(DNAN)为载体的熔铸炸药占90%以上[2-3]。

图1 异呋咱含能化合物的结构Fig.1 Structures of isofurazano energetic compounds
随着武器工业的发展,武器系统的小型化、高能量特性、高安全性已经成为目前军用炸药的具体技术指标,也成为研究者关注的焦点。TNT基熔铸炸药存在能量水平低、热感度高、毒性大、渗油等缺点,严重影响了其在生产、使用、运输和存储过程中的安全性;DNAN的撞击、摩擦和热感度都低于TNT,但是其密度和能量却不及TNT[4]。二者均不能满足现代武器系统作战能力的要求,因此需要寻找高能钝感的新型熔铸炸药载体,用以替代TNT和DNAN. 大量的实践经验表明[5-7],通过分子设计途径,选取适当的取代基团、构建共轭桥联结构和氮杂环结构单元,可以获得符合熔铸载体要求的新型含能化合物。
呋咱环作为一类重要的共轭氮杂环,具有高氮低氢、高生成焓、高能量和一定的芳香性等特点,以呋咱为基的大多数衍生物均具有较低的熔点[8-9],然而由于呋咱化合物机械感度较高、热稳定性差,限制了其作为含能组分在熔铸炸药载体和炸药中的应用[10-11]。异呋咱(1,2,4-噁二唑)作为呋咱的同分异构体,由于其环内氮氧原子分布更加均匀,使得环结构更加稳定,从一定程度上降低了以其为结构单元形成化合物的感度。由于异呋咱与呋咱结构的相似性,以异呋咱环为结构单元构成的化合物可能成为候选的高能化合物。
为了研究异呋咱含能化合物作为新型高能熔铸炸药载体的可能性,本文以异呋咱为构建单元,以偶氮键、C-C键为桥联结构,以硝基、氨基、硝酸酯基为含能取代基,设计了3,4-二硝基异呋咱(A1)、2,2’-二硝基偶氮双异呋咱(A2)、3-硝基呋咱基-5-硝基异呋咱(A3)、3-硝基异呋咱基-5-硝基异呋咱(B1)、3-氨基异呋咱基-5-硝基异呋咱(B2)、3-硝酸酯基异呋咱基-5-硝酸酯基异呋咱(B3)和3,5-二硝基呋咱基氧化异呋咱(C)7种异呋咱化合物(见图1),利用量子化学中的密度泛函理论对所设计化合物的几何构型、密度、生成焓、爆轰参数、熔点、热稳定性、静电势和撞击感度等进行了理论研究,并根据理论计算结果,从7种异呋咱化合物中筛选出符合要求的化合物,作为潜在的新型熔铸炸药载体。本文在研究设计化合物的同时也对文献[12]报道的3,5-二硝基呋咱基异呋咱(DNFO)和3-硝基呋咱基-5-氨基呋咱基异呋咱(ANFO)的能量性能和安全水平进行了理论预估。
1 计算方法

原子电荷是表征化学体系中电荷分布的最简单、最直观的描述形式之一。Mulliken方法[16]中的原子电荷定义如下:
(1)
式中:ZA为原子A的核电荷数;ηi为轨道i占据数;Θm,i表示分子轨道i中基函数m所占的成分。本文采用该方法获得B3LYP/6-311G** 基组水平下部分化合物的原子电荷分布。
含能化合物的密度是直接影响爆轰性能的重要参数之一。本文在B3LYP/6-31G** 基组水平下采用Monte Carlo法[17]计算了0.001 a.u.的电子密度等值面定义的目标化合物分子体积Vm,并利用Rice等[18]提出的公式预测化合物的理论密度ρ:
(2)
式中:M为摩尔质量。


(3)
(4)
基于目标化合物的组成、密度和固相生成焓,运用Kamlet-Jacobs[24]公式计算其理论爆速D和理论爆压p:
(5)
(6)

考察具有最小键级化学键(即最弱键)的键离解能EBDE,是判断含能化合物热稳定性的常用方法之一。对7种异呋咱化合物以及ANFO、DNFO的优化构型进行自然键级轨道(NBO)分析,可得到最弱键的键级,通过(7)式和(8)式计算最弱键的键离解能EBDE值[25]并进行零点能EZPE校正,从而获得校正后的键离解能EBDE*如下:
EBDE=E(R)+E(NO2)-E(R-NO2),
(7)
EBDE*=EBDE+EZPE,
(8)
式中:E(R)表示化合物断裂一个硝基后的总能量;E(NO2)表示硝基的总能量;E(R-NO2)表示化合物的总能量。
Pospisil等[26]认为,分子表面静电势与含能化合物的撞击感度有密切的联系,并提出了撞击感度预测公式。本文在B3PW91/6-31G** 基组水平下绘制了部分化合物的立体静电势分布图,并采用如下经验关系式预测化合物的撞击感度H50(落锤质量为2.5 kg):
(9)
基于基团贡献理论,采用Keshavarz[27]提出的(10)式,对目标化合物的熔点Tm进行预估:
Tm=326.9+5.524a+2.646b+14.60c-2.130d+
101.2Tln,m-68.08TDe,m,
(10)
式中:a、b、c和d分别为C、H、O、N原子的个数;Tln,m和TDe,m分别表示结构单元和硝基、氨基、甲基等取代基团的不同位置以及数量关系对熔点的贡献值。
2 结果与讨论
2.1 几何构型
在B3LYP/6-311G** 基组水平下7种目标化合物的稳定构型如图3(a)~图3(g)所示。由图3可知,所有分子优化后的构型均无虚频,是相对稳定的结构。本文仅仅列出了化合物A2、C的键长和NBO键级数据(见表1)。

图2 异呋咱含能化合物的等键反应Fig.2 Designed isodesmic reactions of isofurazano energetic compounds
异呋咱环和二异环呋咱衍生物(除A3和B3外)的所有原子均处于一个平面上,均为反式构型,其中异呋咱环内各键键长均在1.289~1.389 Å范围,介于正常双键(1.22 Å)和单键(1.47 Å)之间,且趋于平均化;环间相连的C—C键键长均约为1.46 Å,低于烷基中C—C键键长(1.54~1.62 Å);环与硝基之间的C—N键键长均在1.46~1.47 Å之间。这一结果表明A2和C两种化合物的所有化学键的键长越短,其键级越大,几何构型的优化是准确的。

图3 几何结构(蓝色表示氮原子、红色表示氧原子、灰色表示碳原子、白色表示氢原子)Fig.3 Geometric configuration (blue: nitrogen atom; red: oxygen atom; gray: carbon atom; white: hydrogen atom)

A2C化学键键长/Å键级化学键键长/Å键级C(3)—N(6)146908835N(1)—O(12)137111124N(1)—O(2)138910203O(12)—N(11)136011331O(2)—C(3)133711134N(11)—C(10)130216198C(3)—N(4)128915965N(1)—C(9)131515923N(4)—C(5)137311916C(10)—N(14)146109082C(5)—N(9)140910906C(2)—N(3)134312856N(10)—N(9)125520242N(3)—O(22)120714787N(10)—C(11)140910906N(3)—O(5)144610193C(11)—N(13)137311916O(5)—C(13)134610562C(11)—N(12)132214872C(13)—N(4)129815515N(12)—O(14)138910203C(2)—N(4)136212321N(13)—C(15)128915965C(2)—C(9)144910489O(14)—C(15)133711134C(13)—C(6)145410756N(16)—C(15)146908835C(17)—N(19)146309848
化合物A2、B1和C的部分原子Mulliken电荷值如表2所示。由表2可知,A2、B1和C的异呋咱环上原子的总Mulliken电荷均在0.27~0.35 e之间,硝基上N原子的Mulliken电荷均在0.35~0.36 e之间,所有氧原子上均为负电荷。A2中原子的Mulliken电荷呈对称分布,与结构的高度对称性相一致。
2.2 密度

2.3 气相和固相生成焓
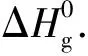

表2 化合物A2、B1和C的部分原子Mulliken电荷

表3 异呋咱化合物和DNFO、ANFO的密度

表4 参考物质在B3LYP/6-311+G** 基组水平下的总能量E0、零点能EZPE、焓校正HT

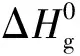


Tab.5 The calculated Δ, ΔsH0 and Δ of isofurazano compounds, ANFO, and DNFO kJ/mol
2.4 爆轰性能与熔点预测
基于7种异呋咱化合物和ANFO、DNFO的密度、固相生成焓,运用K-J方程预测了其爆轰参数,同时计算了目标化合物氧平衡;基于基团贡献理论预估了化合物的熔点[27]。表6中列出了目标化合物的密度、固相生成焓、氧平衡、爆轰参数和熔点预测值;ANFO和DNFO的熔点值取自文献[12]。
从表6中可知,随着异呋咱环的增加,化合物的爆速呈显著上升趋势(A1→B1→C);同时,取代基均为硝基的多环呋咱化合物普遍具有很高的爆速(A2、A3、B1、C和DNFO的爆速均在9.3 km/s以上),由于上述4种化合物均具有较高的正生成焓值和密度,在爆轰过程中放热量巨大,更多的热能转换为爆轰能量,导致爆速很高。取代基为硝基和氨基的多环呋咱化合物(B2和ANFO)相对于硝基多环呋咱化合物,其密度、生成焓和爆速显著下降,表明氨基会降低呋咱类化合物的能量水平。以上分析表明:异呋咱环能有效提高含能化合物的能量性能;硝基对能量的贡献显著高于氨基和硝酸酯基。除A1外,所设计的其余6种化合物熔点均在80~118 ℃之间,符合熔铸炸药的连续相熔点要求。

表6 异呋咱含能化合物和ANFO,DNFO的密度、固相生成焓、氧平衡、爆轰参数和熔点
2.5 热稳定性
7种异呋咱含能化合物以及ANFO、DNFO在B3LYP/6-311+G** 基组水平下最弱化学键的键级和经零点能校正后的键离解能EBDE*[25]计算结果见表7.
由表7可见,异呋咱化合物中的最弱键均为与硝基直接相连的化学键C—N(O2)和(C)O—N(O2)。根据肖鹤鸣等[31]提出的判断高能量密度化合物热稳定性的定量标准:若EBDE*>120 kJ/mol,则认为满足化合物热稳定性要求。除B3外,所有化合物的EBDE*均大于240 kJ/mol,且高于环四亚甲基四硝胺(168 kJ/mol[32]),均具有良好的热稳定性。

表7 异呋咱化合物的最弱化学键的键级和键离解能EBDE*

2.6 静电势与撞击感度
分子的静电势图可以形象地表征分子表面静电势的分布情况。本文绘制了化合物在0.002 a.u.的电子密度等值面上的立体静电势分布图(图4以A2和C为例)。从图4中可以看出:A2的静电势分布高度对称;C中的正静电势主要分布于呋咱基异呋咱环骨架上,负静电势分布于硝基的氧原子附近,与其原子电荷分布相一致。

图4 A2和C的静电势分布Fig.4 Electrostatic potential distribution of A2 and C
利用Multiwfn 3.3.9程序,对7种异呋咱化合物和ANFO、DNFO进行静电势统计分析,获得化合物的部分静电势参数,根据静电势经验关系公式((9)式)计算异呋咱化合物的撞击感度,以特性落高值H50表示,其结果见表8.

表8 异呋咱化合物和DNFO、ANFO的静电势参数和撞击感度
2.7 优选异呋咱含能化合物及其综合性能对比
基于理论计算结果,从7种化合物中筛选出A2和C两种化合物。表9列出了A2和C以及常见熔铸炸药的综合性能参数,其中TNT、DNAN和1,3,3-三硝基氮杂环丁烷(TNAZ)的晶体密度、生成焓和熔点值均取自文献[4,35-36],爆轰参数和撞击感度均为计算值(计算方法与本文相同)。为了佐证本文计算的可靠性,计算了3,4-二硝基呋咱基氧化呋咱(DNTF)的性能参数,同时附上了DNTF的实验数据[37],以供对比(表9中括号内为实验值)。
从表9可知:DNTF的性能参数计算值与实验值误差均在5%以下,说明本文的计算方法是可靠的。A2和C的密度均在1.9 g/cm3以上,固相生成焓和爆速均高于TNT、DNAN和TNAZ,接近DNTF;C的撞击感度高于TNAZ、低于DNTF,A2的撞击感度则与DNTF相当;A2的熔点为80 ℃,处于理想的熔点范围(80~90 ℃)[4];C的熔点接近熔点范围上限(120 ℃),但是由于C作为DNTF的同分异构体,在相容性上具有高度一致性,同时C中的异呋咱环具有一定的芳香性,故C可以与TNT形成低共熔物(DNTF可与TNT形成不同熔点的低共熔物)[37]。综上所述可知,选择A2和C作为合成新型熔铸炸药载体。

表9 优选的两种异呋咱化合物和常见熔铸炸药的综合性能参数表
3 结论
1) 随着异呋咱环数的增加,化合物的密度、生成焓和熔点均增加,撞击感度呈轻微降低趋势;硝基异呋咱化合物熔点普遍低于氨基和硝酸酯基异呋咱化合物。在异呋咱环上引入硝基,在提升能量水平的同时也会降低熔点。除硝酸酯基异呋咱化合物外,其余化合物均满足高能量密度化合物的热稳定性要求。

3) 基于理论计算结果,筛选出A2和C两种化合物,其熔点分别为80 ℃和118 ℃,特性落高分别为20 cm和23 cm,能量接近DNTF,是极具应用潜力的高能量熔铸炸药载体。ANFO作为一种新型钝感熔铸炸药载体,应积极开展其放大合成工艺和以ANFO为基的熔铸炸药配方研究。
)
[1] 张超, 张晓宏, 马亮, 等. LLM-105研究新进展[J]. 科学技术与工程, 2015, 15(23):75-86.
ZHANG Chao, ZHANG Xiao-hong, MA Liang, et al. The new progress of 2,6-diamino-3,5-dinitro pyrazine-1 oxide[J]. Science Technology and Engineering, 2015, 15(23): 75-86. (in Chinese)
[2] 张光全, 黄明. 低熔点钝感高能炸药(IHE)的发展探讨[J]. 含能材料, 2015, 23(2):103-105.
ZHANG Guang-quan, HUANG Ming. Discussion of research on the low melting insensitive high explosive(IHE)[J].Chinese Journal of Energetic Materials, 2015, 23(2):103-105. (in Chinese)
[3] 郑保辉, 罗观, 舒远杰, 等. 熔铸炸药研究现状与发展趋势[J]. 化工进展, 2013, 32(6):1341-1343.
ZHENG Bao-hui, LUO Guan, SHU Yuan-jie, et al. Research status and prospect of melt-cast explosive[J]. Chemical Industry and Engineering Progress, 2013, 32(6):1341-1343. (in Chinese)
[4] 谭彦威, 刘玉存, 杨宗伟, 等. 熔铸炸药的研究进展[J]. 山东化工, 2011, 40(5):22-24.
TAN Yan-wei, LIU Yu-cun, YANG Zong-wei, et al. The current research of the melt-cast explosive[J]. Shandong Chemical Industry, 2011, 40(5):22-24. (in Chinese)
[5] 黄辉. 钝感高能炸药研究进展[J].中国工程物理研究院科技年报, 2013(1):3-8.
HUANG Hui. Research on the progress of insensitive high explosives[J]. Annual Report of China Academy of Engineering Physics, 2013(1):3-8. (in Chinese)
[6] 李云路, 刘田英, 曹端林, 等. 四硝基吡咯及其衍生物结构与性能的理论研究[J]. 含能材料, 2017, 25(4):291-297.
LI Yun-lu, LIU Tian-ying, CAO Duan-lin, et al.Theoretical study on structure and properties of tetranitropyrrole and its derivatives[J]. Chinese Journal of Energetic Materials, 2017, 25(4):291-297. (in Chinese)
[7] Wang K, Shu Y J, Liu N, et al. Theoretical studies on structure and performance of [1,2,5]-oxadiazolo-[3,4-d]-pyridazine-based derivatives[J]. Journal of Physical Organic Chemistry, 2017, 30(1):2-9.
[8] 林智辉, 高莉, 李敏霞, 等. 几种呋咱类含能化合物的合成、热行为及理论爆轰性能预估[J]. 火炸药学报, 2014,37(3):6-19.
LIN Zhi-hui, GAO Li, LI Min-xia, et al. Synthesis, thermal behavior and prediction of theoretical detonation performance for some energetic compounds derived from furazan[J]. Chinese Journal of Explosives & Propellants, 2014, 37(3):6-19. (in Chinese)
[9] 张家荣, 毕福强, 王伯周, 等.α-多硝甲基氧化偶氮含能化合物合成研究进展[J]. 火炸药学报, 2016, 39(6):12-17.
ZHANG Jia-rong, BI Fu-qiang, WANG Bo-zhou, et al. Research progress on the synthesis of energetic compounds based onα-polynitromethyl-azoxy moieties[J]. Chinese Journal of Explosives & Propellants, 2016, 39(6):12-17. (in Chinese)
[10] 李云路, 薛梅, 王建龙, 等. 多硝基呋咱类含能化合物的合成研究进展[J]. 有机化学, 2016, 36(7):1528-1538.
LI Yun-lu, XUE Mei, WANG Jian-long, et al. Advances in the synthesis of poly-nitro furazans[J]. Chinese Journal of Organic Chemistry, 2016, 36(7):1528-1538. (in Chinese)
[11] 张德雄, 张衍, 王琦. 呋咱系列高能量密度材料的发展[J]. 固体火箭技术, 2004, 27(1):32-36.
ZHANG De-xiong, ZHANG Yan, WANG Qi. Advances in high energy density matter of furazan series[J]. Journal of Solid Rocket Technology, 2004, 27(1):32-36. (in Chinese)
[12] Pagoria P F, Zhang M X. Melt-castable energetic compounds comprising oxadiazoles and methods of production thereof: US, US8580054[P]. 2013-10-10.
[13] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian[CP]. Wallingford, CT,US:Gaussian Inc., 2013.
[14] Beck A D. Density-functional thermochemistry.Ⅲ. The role of exact exchange[J]. Journal of Chemical Physics, 1993, 98(7):5648-5652.
[15] Lu T, Chen F W. Multiwfn: a multifunctional wave-function analyzer[J].Journal of Computational Chemistry, 2012, 33(5):580-592.
[16] 卢天, 陈飞武. 原子电荷计算方法的对比[J]. 物理化学学报, 2012, 28(1):1-18.
LU Tian, CHEN Fei-wu. Comparison of computational methods for atomic charges[J]. Acta Physico-Chimica Sinica, 2012, 28(1):1-18. (in Chinese)
[17] 林梦海. 量子化学计算方法与应用[M]. 北京:科学出版社, 2004.
LIN Meng-hai. Computational methods and applications of quantum chemistry[M]. Beijing: Science Press, 2004. (in Chinese)
[18] Rice B M, Byrd E F. Evaluation of electrostatic descriptors for predicting crystalline density[J]. Journal of Computational Chemistry, 2013, 34(25):2146-2151.
[19] 卑凤利, 张兴明, 陈海群, 等. 高能密度材料1,3,4-噁二唑衍生物的分子设计[J]. 南京理工大学学报, 2015, 39(2):246-252.
BEI Feng-li, ZHANG Xing-ming, CHEN Hai-qun, et al. Molecular design of 1,3,4-oxadiazole-based high energy density material[J]. Journal of Nanjing University of Science and Technology, 2015, 39(2):246-252. (in Chinese)
[20] Wei T, Zhu W H, Zhang X W, et al. Molecular design of 1,2,4,5-tetrazine-based high-energy density materials[J]. Journal of Physical Chemistry A, 2009, 113(33):9404-9412.
[21] Wei T, Zhu W H, Xiao H M, et al. DFT study on energetic tetrazolo-[1,5-b]-1,2,4,5-tetrazine and 1,2,4-triazolo-[4,3-b]-1,2,4,5-tetrazine derivatives[J]. Journal of Hazardous Materials, 2010, 179(1):581-590.
[22] 肖鹤鸣. 高能化合物的结构和性质[M]. 北京:国防工业出版社,2004.
XIAO He-ming. Structures and properties of energetic compounds[M]. Beijing: National Defense Industry Press, 2004. (in Chinese)
[23] Byrd E F C, Rice B M. Improved prediction of heats of formation of energetic materials using quantum mechanical calculations[J]. Journal of Physical Chemistry A, 2006, 110(3):1005-1013.
[24] Kamlet M J, Jacobs S J. Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives[J]. Journal of Chemical Physics, 1968, 48(1):23-25.
[25] Li Y L, Wu J P, Cao D L, et al. Theoretical study on the effects of hydrogen-bonding and molecule-cation interactions on the sensitivity of HMX[J]. Journal of Physical Chemistry A, 2016, 120(42):8444-8449.
[26] Pospisil M, Vavra P, Concha M C, et al. A possible crystal volume factor in the impact sensitivities of some energetic compounds[J].Journal of Molecular Modeling, 2010, 16(5): 895-901.
[27] Keshavarz M H. A new computer code for prediction of enthalpy of fusion and melting point of energetic materials[J]. Propellants Explosives Pyrotechnics, 2015, 40(1):150-155.
[28] 张婧婧, 高洪伟, 卫涛, 等. 高能量密度材料3,3’-偶氮-1,2,4,5-四嗪衍生物的分子设计[J]. 物理化学学报, 2010, 26(12):3337-3344.
ZHANG Jing-jing, GAO Hong-wei, WEI Tao, et al. Molecular design of 3,3’-azobis-1,2,4,5-tetrazine-based high-energy density materials[J]. Acta Physico-Chimica Sinica, 2010, 26(12):3337-3344. (in Chinese)
[29] Klapötke T M, Stierstorfer J, Mayr N. Maximum compaction of ionic organic explosives: bis(hydroxylammonium) 5,5’-dinitromethl-3,3’-bi(1,2,4-oxadiazolate) and its derivatives[J]. Chemistry-A European Journal, 2014, 20(5):1410-1417.
[30] Rashid M A M, Cho S G. Heat of formation predictions of various nitro-substituted azoles by G4MP2-SFM scheme[J]. Theoretical Chemistry Accounts, 2015, 134(11):1-11.
[31] 肖鹤鸣, 许晓娟, 邱玲. 高能量密度材料的理论设计[M].北京:科学出版社, 2008.
XIAO He-ming, XU Xiao-juan, QIU Ling. Theoretical design for high energetic materials[M]. Beijing: Science Press, 2008. (in Chinese)
[32] Zhang X W, Zhu W H, Xiao H M. Comparative theoretical studies of energetic substituted carbon- and nitrogen-bridged difurazans[J]. Journal of Physical Chemistry A, 2010, 114(1):611-612.
[33] 朱敏. 若干弱相互作用体系的结构和性质的理论研究[D]. 石家庄:河北师范大学, 2012:11-20.
ZHU Min. Theoretical studies on structures and properties of some weak interactional systems[D]. Shijiazhuang:Hebei Normal University, 2012:11-20. (in Chinese)
[34] Hammerl A, Klapötke T M, Nöth H, et al. Synthesis, structure, molecular orbital and valence bond calculations for tetrazole azide, CHN7[J]. Propellants Explosives Pyrotechnics, 2003, 28(4):165-173.
[35] 张光全, 董海山. 2,4-二硝基苯甲醚为基熔铸炸药的研究进展[J]. 含能材料, 2010, 18(5):604-609.
ZHANG Guang-quan, DONG Hai-shan. Review on melt-castable explosives based on 2,4-dinitroanisole[J]. Chinese Journal of Energetic Materials, 2010,18(5):604-609.(in Chinese)
[36] 刘晶如, 杨寅, 辛伟. 含1,3,3-三硝基氮杂环丁烷(TNAZ)推进剂能量特性计算研究[J]. 固体火箭技术, 2009,32(3):318-322.
LIU Jing-ru, YANG Yin, XIN Wei. Computational investigation of energy characteristics of propellant containing 1,3,3-trinitroazetidine(TNAZ)[J]. Journal of Solid Rocket Technology, 2009, 32(3):318-322. (in Chinese)
[37] 胡焕性, 张志忠, 赵凤起, 等. 高能量密度材料3,4-二硝基呋咱基氧化呋咱性能及应用研究[J]. 兵工学报, 2004,25(2):155-158.
HU Huan-xing, ZHANG Zhi-zhong, ZHAO Feng-qi, et al. A study on the properties and application of high energy density material DNTF[J]. Acta Armamentarii, 2004, 25(2):155-158. (in Chinese)
猜你喜欢
天津建设科技(2022年2期)2022-05-05
动物营养学报(2022年1期)2022-02-20
科技信息·学术版(2021年20期)2021-11-02
陶瓷学报(2021年1期)2021-04-13
火炸药学报(2020年5期)2020-10-27
固体火箭技术(2019年4期)2019-09-10
火工品(2019年3期)2019-09-02
火炸药学报(2018年4期)2018-09-01
江苏农业科学(2017年10期)2017-07-21
今日农药(2014年7期)2014-09-15
