
谷胱甘肽合成酶缺乏症2例基因分析及文献回顾
2017-12-08 05:46:47汪治华
中国妇幼健康研究 2017年11期
刘 超,汪治华
(西安市儿童医院内分泌遗传代谢科,陕西 西安 710003)
谷胱甘肽合成酶缺乏症2例基因分析及文献回顾
刘 超,汪治华
(西安市儿童医院内分泌遗传代谢科,陕西 西安 710003)
目的探讨谷胱甘肽合成酶缺乏症(GSSD)的临床及遗传学特点。方法对2014年1至12月西安市儿童医院内分泌遗传代谢科临床诊断的2例5-羟脯氨酸尿症的患儿进行临床特点分析,采用目标序列捕获测序方法,对患儿进行谷胱甘肽合成酶(GSS)基因分析,再经聚合酶链式反应(PCR)对高危突变基因进行验证,最终确诊为GSSD;并进行相关临床特点总结及基因学分析。结果GSSD临床表现为代谢性酸中毒、黄疸、溶血性贫血,GSS基因检测出致病突变,其中1例为复合杂合突变(E5 c.491Ggt;A 和 E10 c.847Cgt;T),1例为纯合突变(E5 c.491Ggt;A)。结论E5 c.491Ggt;A基因突变可能为GSSD热点基因突变。
5-羟脯氨酸酶;谷胱甘肽合成酶;5-羟脯氨酸尿症;基因分析;基因突变
谷胱甘肽是一种由谷氨酸、半胱氨酸、甘氨酸组成的三肽,参与体内游离基的清除、氧化还原反应、脱氧核苷酸的形成、生物异源性物质的代谢以及氨基酸的转运,其生物合成及代谢分解均在γ-谷胺酰循环中完成。在6种γ-谷胺酰循环酶中,已有4种酶的遗传性缺陷见诸报道。其中,谷胱甘肽合成酶(glutathione synthetase,GSS)和5-羟脯氨酸酶(5-oxoprolinase,OPLAH)的缺陷,是造成大量5-羟脯氨酸蓄积的重要原因,致使临床上出现5-羟脯氨酸尿症(又称为焦谷氨酸尿症)。因此,根据致病基因不同,5-羟脯氨酸尿症可分为谷胱甘肽合成酶缺乏症(glutathione synthetase deficiency,GSSD)和5-羟脯氨酸酶缺乏症两种类型。
本研究选取2014年1至12月西安市儿童医院2例临床诊断5-羟脯氨酸尿症的患儿家系,提取全血基因组DNA,依据5-羟脯氨酸尿症致病基因GSS的基因组序列,分析测序结果寻找突变位点,结合相关文献,以期提高临床对GSS缺乏症的认识。
1临床资料
2例临床诊断为5-羟脯氨酸尿症的患儿体重分别为3.7kg、3.4kg,出生后不久即发病,均表现为气促、精神差,无抽搐发作,查血气分析示代谢性酸中毒,反复给予碳酸氢钠治疗后,酸中毒不易纠正。血常规提示中度贫血,血乳酸、血氨、血同型半胱氨酸检测及血氨基酸、酰基肉碱检测未见明显异常。尿有机酸检测提示:5-羟脯氨酸分别为316.4μmol/L及210.5μmol /L(正常参考值:0~7.6μmol/L)。临床诊断为5-羟脯氨酸尿症。
经患儿家属同意后,留取患儿及父母外周血标本,提取全血基因组DNA,依据GSS的基因组序列,设计引物,利用聚合酶链式反应(polymerase chain reaction,PCR)扩增该基因编码区的全部外显子,PCR产物直接测序后分析测序结果寻找突变位点。
2结果
经与NCBI数据库中正常人GSS基因序列进行比对,证实病例1 患儿GSS基因的第5号外显子在c.491Ggt;A和第10号外显子在c.847Cgt;T各有一个致病突变位点,其类型均为杂合错义突变;病例2 患儿GSS基因的第5号外显子在c.491Ggt;A有一个纯合错义突变,见图1。2例患儿最终均被确诊为GSSD。难以纠正的代谢性酸中毒、黄疸、溶血性贫血、尿5-羟脯氨酸大量排泄是其重要的临床特征。





注:A为正常对照序列;B为病例1 外显子5(E5)c.491Ggt;A(p.Arg164Gln)错义突变,外显子10(E10)c为847Cgt;T(p.Arg283Cys)错义突变;C为病例1父亲外显子5(E5)c.491Ggt;A(p.Arg164Gln)错义突变,外显子10(E10)正常;D为病例1母亲 外显子5(E5)正常,外显子10(E10)c.847Cgt;T(p.Arg283Cys)错义突变;E为病例2 外显子5(E5)c.491Ggt;A(p.Arg164Gln)纯合错义突变。
图1基因测序图
Fig.1 Gene sequencing map
3讨论
3.1 γ-谷胺酰循环需多种酶共同参与完成
谷胱甘肽(γ-谷氨酰半胱氨酰甘氨酸)的生物合成,是由γ-谷氨酰半胱氨酸合成酶和谷胱甘肽合成酶催化的。起始的降解步骤由γ-谷氨酰转肽酶催化,它将γ-谷氨酰基转移至受体,例如氨基酸,就生成γ-谷氨酰氨基酸。谷胱甘肽合成酶是典型的γ-谷氨酰环化转移酶的底物,该酶催化γ-谷氨酰剩余物如5-羟脯氨酸的释放,而5-羟脯氨酸则在5-羟脯氨酸酶的作用下转化为谷氨酸,见图2。谷胱甘肽起着γ-谷氨酰半胱氨酸合成酶反馈性抑制剂的作用。
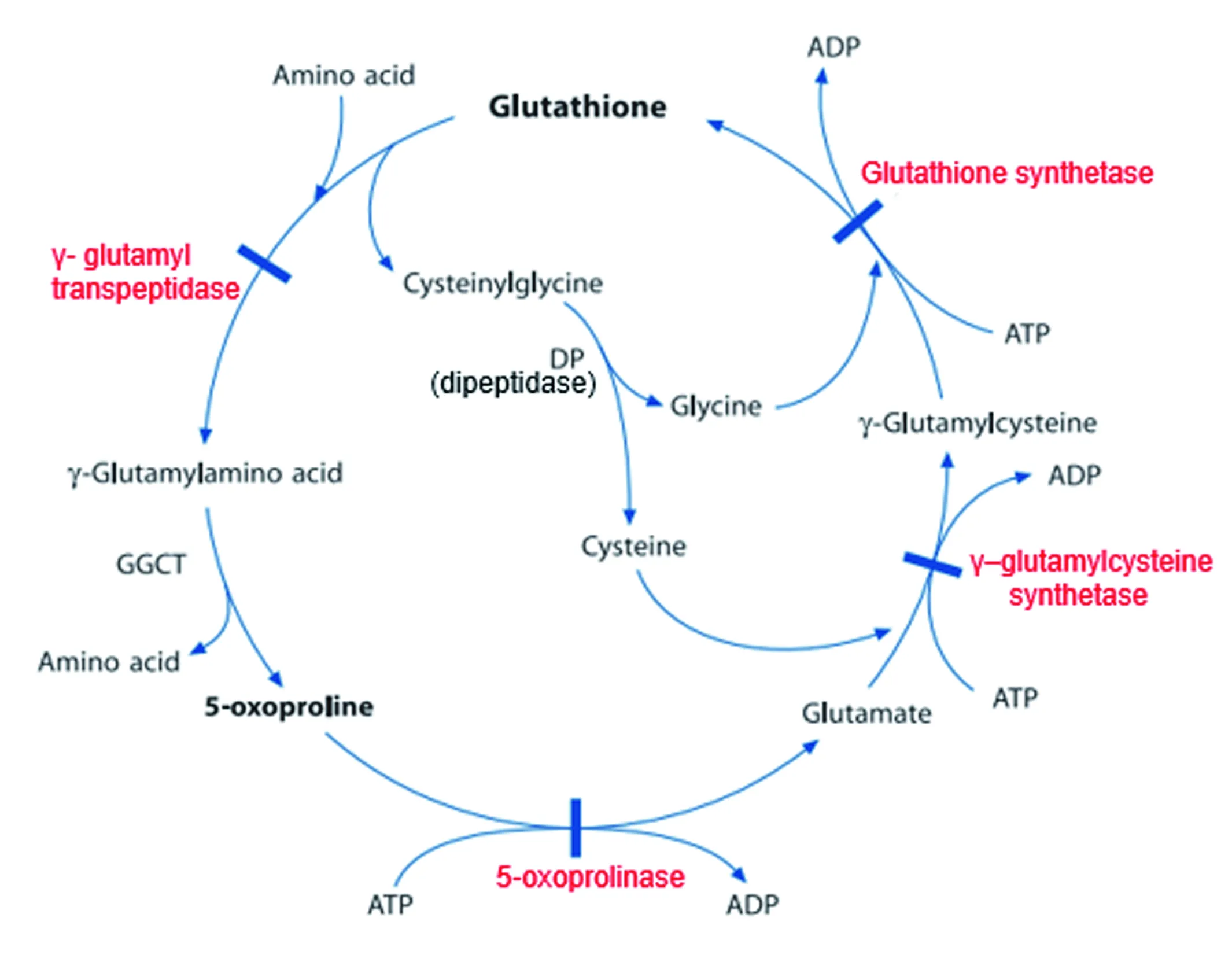
注:GGCT=γ-谷氨酰环化转移酶;DP=二肽酶
图2 γ-谷胺酰循环图
Fig.2 Gamma-glutamyl cyclic graph
在γ-谷胺酰循环中,可存在4种酶缺陷,其中仅以5-羟脯氨酸酶和谷胱甘肽合成酶缺陷以5-羟脯氨酸(焦谷氨酸)过量为标记。本研究中,2例患者尿有机酸检测均表现为5-羟脯氨酸尿症。
3.2谷胱甘肽合成酶缺乏症的重要临床特征
GSSD是一种极为罕见的常染色体隐性遗传病,大多数患者于新生儿期被发现,分为两种:一种只有红细胞的改变,患者的唯一症状就是溶血性贫血;另一种为全身性GSSD,其症状包括新生儿严重的难易纠正的代谢性酸中毒、溶血性贫血和新生儿高胆红素血症,最终出现进行性的神经系统症状(如痉挛、抽搐、智力低下等)。Ellionor等人于2007年报道发现尿中5-羟脯氨酸显著增高,红细胞或成纤维细胞谷胱甘肽合成酶活性降低,仅为正常人群10%左右,而杂合子携带者酶活性约为正常人的50%~70%。Verma等[1]及Milosevic等[2]研究指出,过量生成5-羟脯氨酸(焦谷氨酸)所造成的高阴离子间隙性代谢性酸中毒是γ-谷氨酰循环反馈性调节缺陷的结果。
全世界报道的GSSD患者大约有70人,来自50余家庭。约25%的GSSD患者因感染和(或)电解质紊乱死于新生儿期。通过其临床表现,可分为3种类型,即轻型、中度、重型。轻型患儿主要有基因突变作用于酶活性的稳定性,有核细胞有足够高的蛋白质周转来弥补这种稳定缺陷。然而,谷胱甘肽对细胞膜完整性至关重要。缺乏成熟红细胞的蛋白质合成会造成孤立的溶血性贫血。中度患儿表现为严重的代谢性酸中毒和尿液中5-羟脯氨酸的大量检出,甚至高达0.5~1.0g·kg-1·d-1。重型患者除上述特征外,会进一步出现神经系统功能损害,包括痉挛、抽搐、智力低下及精神运动发育障碍等。本研究中,2例患儿均为中度,尚未出现中枢神经系统损害。此外,Ristoff 等曾在2007年研究证实,GSSD可引起粒细胞功能缺陷而致使细菌感染的易感性增加,一些病例会出现渐进性视网膜营养不良和黄斑色素沉着。该病还被认为和产前大脑出血、半胱氨酸、白三烯水平低下及多发畸形有关。
3.3谷胱甘肽合成酶缺乏症的诊断及鉴别诊断
在γ-谷氨酰循环障碍中引起尿液中5-羟脯氨酸含量增高的代谢缺陷,除谷胱甘肽合成酶缺陷外,5-羟脯氨酸酶缺乏症患者尿液中亦可检出大量5-羟脯氨酸。但该病不出现酸碱平衡紊乱及溶血性贫血。
在诊断的过程中,除GSSD和5-羟脯氨酸酶缺乏症外,还应注意以下因素引起尿液中5-羟脯氨酸升高:①饮食,某些婴儿配方奶含有酪蛋白,经加热后,可转化为5-羟脯氨酸;在番茄经由化学处理生产番茄汁的过程中,可将谷氨酰胺转化为5-羟脯氨酸。②严重的烧伤及Stevens-Johnson综合征的患者,包含大量5-羟脯氨酸的一些蛋白质如纤维蛋白原和胶原蛋白等代谢增加。③除γ-谷氨酰循环障碍以外的先天遗传代谢缺陷疾病,包括X-连锁的鸟氨酸氨甲酰转移酶缺乏症、尿素循环障碍、酪氨酸血症。④在肝脏、肾脏等重要器官,若缺乏ATP,5-羟脯氨酸将不能转化为谷氨酸,可导致5-羟脯氨酸尿症。⑤高胱氨酸尿症。⑥药物代谢如扑热息痛、氨己烯酸和抗生素(氟氯西林)等。⑦早产儿,尤其在极早产儿中,可出现一过性的5-羟脯氨酸尿症。⑧营养不良、孕期限制甘氨酸的摄入。⑨胱氨酸肾病。胱氨酸肾病患者出现5-羟脯氨酸尿症的原因可能是半胱氨酸的可用性降低,导致γ-谷氨酰循环的继发性损伤。
GSSD临床表现为难以纠正的代谢性酸中毒、溶血性贫血、黄疸、尿5-羟脯氨酸大量排泄,在排除其他继发性因素所致尿5-羟脯氨酸大量排泄后,应考虑5-羟脯氨酸尿症,为进一步明确诊断,需行基因检测。截至2016年,国内仅由彭薇等[3]、Li等[4]报道筛查出5-羟脯氨酸尿症7例,其中3例经基因检测确诊为GSSD。随着检测技术的进步,新的基因检测方法将为患者诊断提供巨大帮助,可指导临床治疗,为产前诊断、遗传咨询提供有力依据。
3.4谷胱甘肽合成酶缺乏症基因学分析及热点基因突变推测
人类谷胱甘肽合成酶是一个由474的氨基酸构成的同型二聚体,它有一条单拷贝基因编码。该基因存在于染色体20q11.2,包括13个外显子,长度约32kb。GSSD是一种罕见的常染色体隐性遗传病,目前已被证实GSSD基因有近30种不同的突变类型。由于等位基因的异质性,GSSD患者间临床表型多样。Njålsson等人在2005年报道41例GSSD,轻型患儿突变类型以656A→G为主;中型患儿突变类型以491G→A,c129+1663A→G,77C→A 为主;重度患儿c.-9+5G→A,847C→T,374G→A为主。本研究及Li等[4]共报道5例患儿,均发现491G→A基因突变,其临床表型与基因型一致。因此,可推测该基因可能为GSSD热点基因突变。
3.5谷胱甘肽合成酶缺乏症的治疗进展
GSSD是需要终身治疗的。治疗包括:纠正酸中毒、口服α-生育酚和甜菜碱、避免服用引起溶血的药物。为了防止出现氧化应激反应,大剂量的维生素C(100mg·kg-1·d-1)和维生素E(100~300mg/d)是必须的[5]。维生素E还可以防止出现粒细胞功能障碍,从而减少急性细菌感染的发生率。Ristoff等在2001年一项针对46例GSSD患者的长期随访研究表明,维生素C和维生素E治疗可能会保护神经系统免受损害。重度GSSD患者早期给予维生素C和维生素E治疗尤其重要。目前没有办法证实这些患者何时会出现神经系统损害,因此,无论GSSD患者严重程度如何,都应早期给予维生素C和维生素E治疗。硒通过合成硒蛋白具有强大的抗氧化特性,目前已有学者应用于GSSD治疗,并取得良好效果[6]。代谢性酸中毒是GSSD重要的临床特征之一,无论是急性发作期或是临床缓解期,都应给予碳酸氢钠纠正酸中毒[7],且疗效值得肯定。所有治疗中应避免在6-磷酸葡萄糖脱氢酶(G6PD)缺陷中引起溶血危象的药物和食物等触发因子。
先前,N-乙酰半胱氨酸被用来作为治疗药物,因其可增加正常人细胞内谷胱甘肽水平。然而,N-乙酰半胱氨酸已经被证实可增加细胞内半胱氨酸的水平,而GSSD患者细胞内半胱氨酸含量已显著升高,高水平的半胱氨酸具有神经毒性,可加重GSSD患者中枢神经系统损害[5]。儿童使用对乙酰氨基酚的有效性及安全性是已经被证实的。一般来说,对乙酰氨基酚的毒性反应出现在年龄比较小的儿童。但是,Tokatli等人在2007年研究指出,GSSD患者即便使用治疗剂量的对乙酰氨基酚仍可能出现毒性反应,诱发肝毒性。因而,GSSD患者不提倡使用N-乙酰半胱氨酸及对乙酰氨基酚,应引起临床重视。
由于目前孕产妇群体对遗传代谢性疾病筛查认知水平较低且主动获知意识差[8],因此,以医院为主开展多渠道、多样化的健康教育工作以提高新生儿遗传代谢病筛查认知水平是很有必要的,这也将大大提高遗传代谢性疾病的检出率。目前本研究2例患者随访结果显示,尿5-羟脯氨酸均得到良好控制,未再出现代谢性酸中毒、溶血性贫血、黄疸,神经系统未见明显异常,精神运动发育较同龄儿无明显落后,临床预后良好。这主要归咎于早期的新生儿遗传代谢性疾病筛查及对5-羟脯氨酸尿症的早期识别、早期诊断、早期治疗。因此,我们呼吁对临床发现不明原因的代谢性酸中毒、溶血性贫血、黄疸,应行尿有机酸检测。若发现尿5-羟脯氨酸水平显著升高,应及时行GSSD相关基因学检测,早期给予大剂量维生素C和维生素E治疗,可获得良好预后。
[1]Verma R,Polsani K R,Wilt J,etal.5-Oxoprolinuria as a cause of high anion gap metabolic acidosis[J].Br J Clin Pharmacol,2012,73(3):489-491.
[2]Milosevic S,Tran K,O’Brien B.A rare cause of high anion gap metabolic acidosis[J]. Intern Med J,2013,43(1):100-101.
[3]彭薇,张万巧,封志纯.气相色谱-质谱法检测遗传代谢性疾病高危患儿[J].临床儿科杂志,2014,32(9):888-891.
[4]Li X,Ding Y,Liu Y,etal.Five Chinese patients with 5-oxoprolinuria due to glutathione synthetase and 5-oxoprolinase deficiencies[J].Brain Dev,2015,37(10):952-959.
[5]Ben Ameur S,Aloulou H,Nasrallah F,etal.Hemolytic anemia and metabolic acidosis: think about glutathione synthetase deficiency[J]. Fetal Pediatr Pathol,2015,34(1):18-20.
[6]Atwal P S,Medina C R,Burrage L C,etal.Nineteen-year follow-up of a patient with severe glutathione synthetase deficiency[J].J Hum Genet,2016,61(7):669-672.
[7]Gündüz M,Ünal Ö,Kavurt S,etal.Clinical findings and effect of sodium hydrogen carbonate in patients with glutathione synthetase deficiency[J].J Pediatr Endocrinol Metab,2016,29(4):481-485.
[8]王蕊,欧明才,孟仙,等.孕产妇对新生儿遗传代谢病筛查认知情况调查[J].中国妇幼健康研究,2014,25(6):1055-1056,1059.
[专业责任编辑:肖延风]
Geneanalysisandliteraturereviewofglutathionesynthetasedeficiencyintwocases
LIU Chao, WANG Zhi-hua
(DepartmentofGeneticMetabolicEndocrine,Xi’anChildren’sHospital,ShaanxiXi’an710003,China)
ObjectiveTo investigate the clinical and genetic characteristics of glutathione synthase deficiency (GSSD).MethodsThe clinical characteristics of 2 infants diagnosed with 5-oxoprolinuria in Xi’an Children’s Hospital were analyzed retrospectively. The glutathione synthetase (GSS) gene was analyzed by target sequencing method. The high-risk mutations genes were verified by polymerase chain reaction (PCR). The final diagnosis was GSSD and relevant clinical features and genetics were analyzed.ResultsThe clinical manifestations of GSSD were metabolic acidosis, jaundice and hemolytic anemia. One case was detected to be compound heterozygous mutation (E5 c.491Ggt;A and E10 c.847Cgt;T) and the other was homozygous mutation (E5 c.491Ggt; A) by GSS gene analysis.ConclusionE5 c.491Ggt; A gene mutation might be an important gene mutation of GSSD.
5-oxoprolinase; glutathione synthetase (GSS); 5-oxoprolinuria; gene analysis; gene mutation
10.3969/j.issn.1673-5293.2017.11.040
R722
A
1673-5293(2017)11-1435-04
2017-07-09
刘 超(1984—),男,主治医师,硕士,主要从事儿童内分泌及遗传代谢性疾病诊疗工作。
汪治华,主任医师。
猜你喜欢
现代食品(2023年21期)2024-01-18 09:52:22
纺织标准与质量(2022年1期)2022-07-12 06:01:22
家庭医学(下半月)(2019年9期)2019-10-12 08:03:56
山东化工(2019年12期)2019-02-16 14:38:58
中国酿造(2018年7期)2018-08-10 02:16:48
家庭用药(2016年9期)2016-12-03 08:25:21
食品安全导刊(2016年15期)2016-03-27 21:26:46
医学研究杂志(2015年3期)2015-06-10 06:41:52
天津医科大学学报(2015年3期)2015-06-05 12:21:49
中国现代医生(2014年12期)2014-06-10 00:52:32
