
香豆素及其衍生物与CHF3的团簇的理论研究
2017-11-01 07:14张郅琨谭会艳张丽娟
山东化工 2017年18期
张郅琨,张 斌,谭会艳,王 跃,张丽娟*
(1.滨州学院 化学化工学院,山东 滨州 256600;2.中海沥青股份有限公司,山东 滨州 256600)
香豆素及其衍生物与CHF3的团簇的理论研究
张郅琨1,张 斌2,谭会艳1,王 跃1,张丽娟1*
(1.滨州学院 化学化工学院,山东 滨州 256600;2.中海沥青股份有限公司,山东 滨州 256600)
采用密度泛函理论中的M06-2X方法,以6-311++G(d,p)为基组,对香豆素分子和7-氟-香豆素分子的单体及其分别与三氟甲烷形成的团簇进行了几何构型优化和振动频率计算。结果表明,香豆素及其衍生物分子上具有不同的团簇结合位点,且氟取代基的存在使团簇结合能降低;应用分子中的原子理论(AIM)证明了团簇中的分子间作用是氢键作用;分析团簇的构型结果显示,在CH...X(X=O, F)型团簇中,CHF3中质子供体C-H键的键长伸长,振动频率蓝移,而在CH…π型团簇中C-H键长和振频的变化没有明显规律。在此基础上,本文采用自然键轨道(NBO)理论对部分体系的氢键形成机理进行了解释,结果表明当重杂化效应占主导时,表现为蓝移氢键。
香豆素;三氟甲烷;量子化学计算;结合能;氢键
香豆素类化合物是具有苯并吡喃环母核的一类天然化合物,具有优良的生理活性和潜在的药用价值,而药物发挥药效的化学本质是药物分子与生物靶分子之间的相互作用,因此在分子水平上研究香豆素类衍生物及其团簇的构型和相互作用能等信息,一方面可以为已有的光谱实验结果提供理论依据,另一方面也对将来探究香豆素类化合物与蛋白质分子作用的药物机理具有一定的指导意义[1]。江欢等[2]采用实验方法研究7-羟基香豆素分别与三种芳香族氨基酸的相互作用,发现分子间存在不同类型的氢键作用。Su等人[3]采用理论计算方法研究了香豆素102(C102)在乙醇(EtOH)溶液中的激发态氢键动力学,结果显示分子间的氢键作用在激发态增强。目前,研究人员对于某些香豆素衍生物的单体进行了实验和理论计算研究[4-7],但对取代基效应对香豆素衍生物团簇的理论计算研究相较少。本文选择香豆素分子(Co)和7-氟香豆素(7F-Co)单体及其与CHF3形成的团簇为研究对象,探究不同团簇构型及其分子间作用的性质和强弱。
1 计算方法
本文使用Gaussian 09程序[8],在M06-2X/ 6-311++G(d,p)计算水平上对两种香豆素衍生物的单体及其与CHF3形成的团簇进行构型优化和振动频率分析,所有计算结果均没有虚频,所得团簇的结合能(BE)均进行了零点能校正。采用AIM 2000程序[9]对所研究的团簇体系进行电子密度拓扑分析;并采用自然键轨道(NBO)理论分析单体和团簇的轨道与电荷,从而对团簇中的氢键性质进行解释,这部分计算采用Gaussian 09自带的NBO 3.1完成。
2 结果与讨论
2.1 团簇几何构型和结合能
图1和2列出了在M06-2X/6-311++G(d,p)计算水平上,优化得到的香豆素(Co)、7-氟-香豆素(7F-Co)分别与CHF3形成的稳定构型(括号内为分子间作用类型),以及及团簇结合能(BE)大小。
如图1所示,Co…CHF3团簇构型中,香豆素分子有主要存在四个结合位点,包括香豆素上羰基O、杂环O原子、苯环上方的π电子云和杂环上方的电子云,总体可分为C-H…O和C-H…π两种作用类型,因为O原子上的孤对电子n(O)作质子受体的能力比较强,故形成C-H…O型的团簇结合能比C-H…π团簇的结合能均要大。其中,构型一中存在两种相互作用,包括C-H…O=C和C-H…O,故所得团簇的结合能也最大,为-6.423 kcal/mol;比较构型三和构型四,对于C-H…π作用为主的团簇构型,在苯环上方要比在杂环上方的结合能大,主要由于苯环上的电子云密度大,其作为质子受体的能力比杂环强。
如图2,对于7F-Co…CHF3团簇,除了上述四个结合位点外,7位的F取代基也可作为新的质子受体,形成CH…F作用的团簇(构型五),但因氟原子作为电子供体能力较弱,所以形成的团簇的结合能只有-2.396 kcal/mol,在五种构型中作用最弱。比较图1和图2可以发现,相同构型情况下(构型一到构型四),7F-Co…CHF3的团簇结合能要比Co…CHF3的结合能小,这主要是因为氟原子作为典型的吸电子取代基,会导致分子主体的电子云密度降低,从而使得分子间作用能减小。
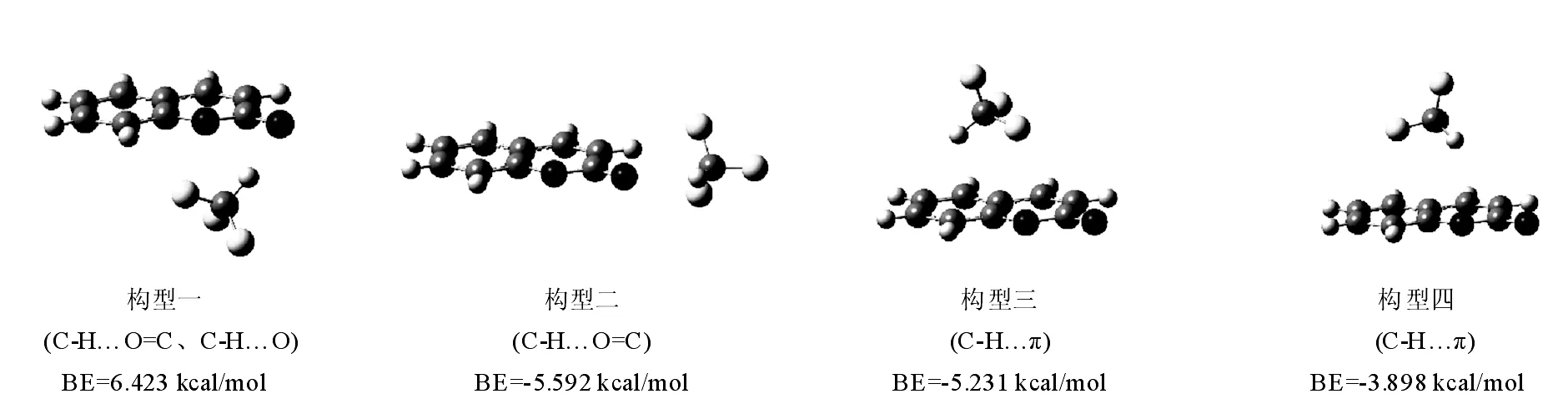
图1 在M06-2X/6-311++G(d,p)水平上优化得到的香豆素和CHF3的团簇的稳定几何构型
Fig.1 The stable geometric structures of coumarin...CHF3cluster optimized at the M06-2X/6-311++G(d,p) level
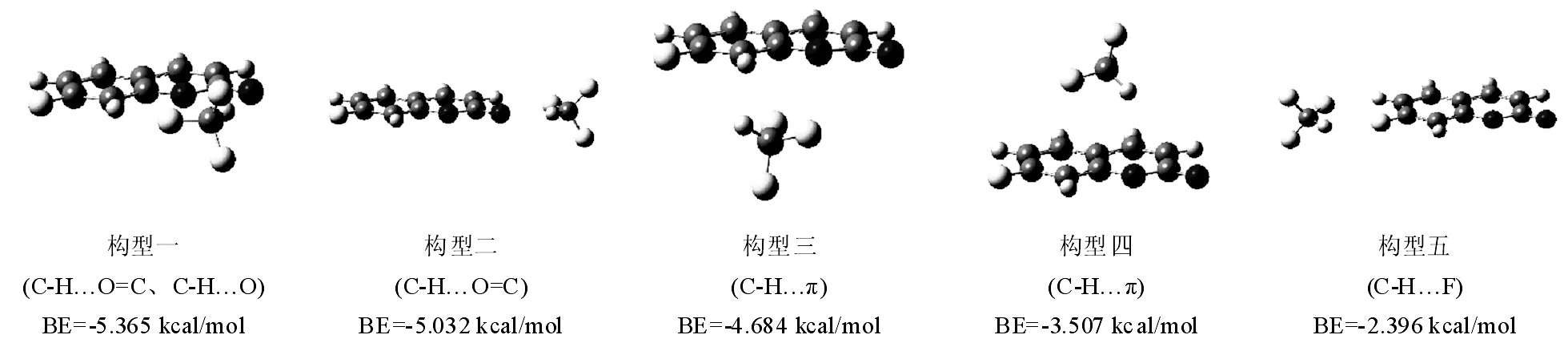
图2 在M06-2X/6-311++G(d,p)水平上优化得到的7-氟香豆素和CHF3的团簇的稳定几何构型
2.2 团簇的电子密度的拓扑分析
基于Bader提出的AIM理论[10]所进行的电子密度拓扑分析是研究氢键的有力工具。对本文所研究的团簇体系,我们在M06-2X/6-311++G(d,p)水平上计算了其电子密度的拓扑性质。Popelier[11]等在AIM理论基础上提出的氢键判断的标准主要有:在H原子与质子受体之间存在键临界点(BCP)与相应的成键路径,且BCP处的电子密度ρ(BCP) 在0.002~0.034 a.u.之间,电子密度的Laplacian值▽2ρ(BCP) 在0.024~0.139 a.u.之间。由此,本文主要分析了BCP即(3,-1)这个点的性质,计算结果如表1所示,根据上述三个标准,可以判定Co…CHF3和7F-Co…CHF3中的分子间作用均为氢键作用。
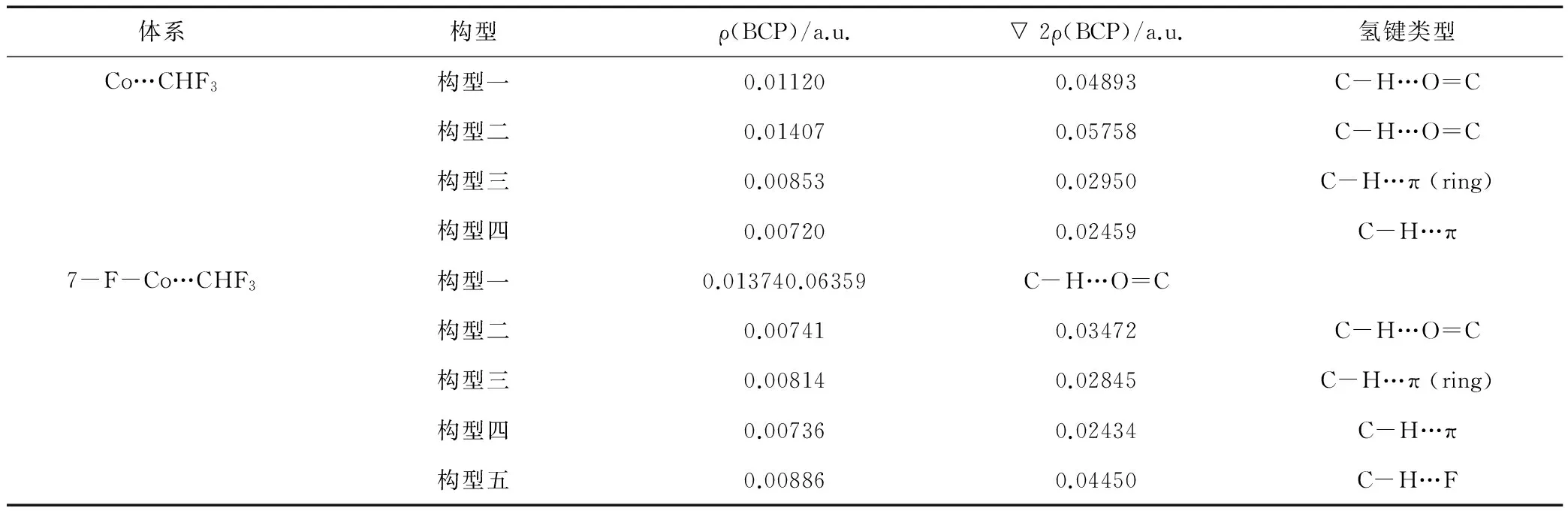
表1 香豆素和7-氟香豆素分别与CHF3形成的团簇的AIM分析
2.3 键长和振动频率分析
表2列出了CHF3单体及其分别与Co和7F-Co形成团簇后对应的C-H键的键长、伸缩振动频率及其变化值。传统氢键(X-H…Y)的典型特征是,形成团簇之后,X-H键长伸长,X-H伸缩振动频率减小(红移)。而对于本研究体系,计算结果显示,对于CH...X(X=O, F)型的团簇,CHF3在参与形成氢键团簇后,其C-H键的键长缩短,伸缩振动频率增大,这种氢键也被称为蓝移氢键[12];并且C-H键长缩短越大,对应的C-H键的伸缩振动频率蓝移也越大,在两种团簇中,均为构型二的C-H键的键长缩短程度最大,对应的C-H键的伸缩振动频率蓝移也最大,分别为77.83 cm-1和72.07 cm-1。而对于在苯环上方的CH...π型团簇,团簇中CHF3的C-H键的键长有较小程度的伸长,但是振动频率却发生了蓝移,这在前人的研究中也有相似的现象出现[13];对于在杂环上方的CH...π型团簇,Co…CHF3中的C-H键同样有较小程度的伸长,振动频率发生蓝移,而7F-Co…CHF3中的C-H键长却有较小程度的伸长,振动频率发生的较小的红移,后者与传统的"红移氢键"的特征吻合。
综上构型分析结果,Co和7F-Co分别与CHF3形成氢键团簇后,质子供体的X-H的键长变化和振动频率变化与传统红移氢键有一定差异,不能用传统的机理进行解释。
2.4 自然键轨道(NBO)分析
为探究团簇体系的构型变化原因,本文采用NBO理论对单体和团簇进行了分析。根据Alabugin等[14]提出的理论,所有类型的氢键都可以用超共轭作用和重杂化作用来进行解释,其中前者会使C-H键伸长,而后者会使C-H键长缩短,两种效应的相对大小决定了C-H键长是伸长还是缩短。
计算结果显示,对于CH...X(X=O, F)型的团簇,O或F上的孤对电子密度几乎没有改变,而CHF3中σ*( C-H)电子密度增加也不明显,孤对电子轨道与σ*( C-H)轨道的超共轭作用能很小,其中 Co…CHF3的构型一和构型二体系,n(O)→σ*( C-H)的超共轭作用能分别只有0.83和0.64 kcal/mol,7F-Co…CHF3的构型一和构型二体系,n(O)→σ*( C-H)的超共轭作用能分别只有0.45和1.70 kcal/mol。另一方面,比较团簇和单体中CHF3的C-H键杂化轨道内s成分的改变,其中 Co…CHF3的构型一和构型二体系,C-H键杂化轨道内s成分的大小由单体的30.52%分别变成31.54%和31.92%;7F-Co…CHF3的构型一和构型二体系,C-H键杂化轨道内s成分的大小由单体的30.52%分别变成31.85%和32.03%。比较两种效应的大小可以看出,对于CH...X(X=O, F)型的团簇,使C-H键缩短的重杂化效应比超共轭效应更占优势,因而表现为键长缩短,振动频率蓝移。而对于CH...π型的团簇,出现了CHF3中C-H伸长,但C-H振动频率发生蓝移的情况,使用该理论不能进行比较好的解释,推测可能是由于团簇中同时存在的其他弱相互作用如分子内超共轭作用的影响造成的,但还需要今后进一步完善相应的理论对其进行解释。
4 结论
本文应用量化计算方法在M06-2X/6-311++G(d,p)的水平上对香豆素和7-氟-香豆素分子分别与CHF3形成的团簇体系进行了研究,结果发现在这些体系中,在不同的结合位点可以形成不同类型的几何构型,其中C-H…O型团簇的结合能比CH…π型团簇的结合能要大。通过对团簇的电子密度的拓扑分析证明了这个分子间相互作用的氢键性质。比较单体和团簇的构型分析发现,不同的氢键团簇的形成对CHF3的键长和振动频率有不同的影响,本文采用NBO理论对部分体系中的氢键进行了解释。
[1] 刘雪峰,夏咏梅,方 云,等.三中香豆素类中药小分子与牛血清蛋白的相互作用[J].化学学报, 2004, 62(16): 1484-1490.
[2] 江 欢,朱燕舞,王 燕,等.7-羟基香豆素与三种芳香族氨基酸作用的荧光光谱研究[J].光谱学与光谱分析, 2013 (08): 2117-2122.
[3] Su J, Tian D X. Strengthening of hydrogen bonded coumarin 102 in ethanol solvent upon photoexcitation[J].New Journal of Chemistry, 2014, 38(02): 568-573.
[4] 靳瑞发,李 杰,孙卫东. 7-甲氧基香豆素-3-甲酰二乙醇胺的电子结构和光谱性质的理论研究[J]. 化学研究与应用, 2009 (07): 950-954.
[5] 贾飞云, 张波, 苏宇. 几种香豆素衍生物电子光谱的理论研究[J]. 计算机与应用化学, 2015 (07): 818-820.
[6] 苏 宇,贾飞云,冉 鸣,等.7-羟基香豆素红外光谱的密度泛函理论研究[J]. 光谱学与光谱分析, 2016 (01): 60-63.
[7] Sert Yusuf, Puttaraju K B, Keskinog Sema,et al. FT-IR and Raman vibrational analysis, B3LYP and M06-2X simulations of 4-bromomethyl-6-tert-butyl-2H-chromen-2-one[J]. Journal of Molecular Structure, 2015, 1079: 194-202.
[8] Frisch M J, Schlegel H B, Suzerai G E, et al. Gaussian 09, Revision D. 01, Gaussian[CP]. Inc: Wallingford, CT, 2004.
[9] Biegler-König F, Schönbohm J, Bayles D. AIM 2000 - A program to analyze and visualize in molecular [J]. Journal of Computational Chemistry, 2001, 22: 545-559.
[10] Bader R F W. Atoms in Molecules[M].New York: Oxford University Press, 1990.
[11] Koch U, Popelier P L A. Characterization of C-H···O hydrogen bonds on the basis of the charge density [J]. Journal of Physical Chemistry, 1995, 99(24): 9747-9754.
[12] Pavel Hobza, Zdenek Havlas. Blue-shifting hydrogen bonds [J]. Chemical Reviews, 2000, 100: 4253-4264.
[13] 倪 杰,黎安勇,闫秀花. HNO(HNS)与分子簇(HF)1≤n≤3形成蓝移氢键的理论研究[J]. 西南大学学报(自然科学版), 2009, 31(01): 49-54.
[14] Igor V Alabugin, Mariappan Manoharan, Scott Peabody, et al. Electronic basis of improper hydrogen bonding: A subtle balance of hyperconjugation and rehybridization [J]. Journal of the American Chemical Society, 2003, 125: 5973-5987.
TheoreticalStudyontheClustersofCoumarinanditsDerivativewithCHF3
ZhangZhikun1,ZhangBin2,TanHuiyan1,WangYue1,WangYue1,ZhangLijuan1*
(1.College of Chemistry & Chemical Engineering, Binzhou University, Binzhou 256600,China;2.China Offshore Co.,Ltd.,Binzhou 256600,Cihna)
In this work, the geometry optimization and vibrational frequency calculations were performed for the monomers of coumarin and 7-fluoro-coumarin and their clusters with fluoroform at the M06-2X/ 6-311++G(d,p) level of theory. The results indicated that there are different binding sites in the molecules, and the binding energies of the clusters decreases due to the fluorine substituent. Atoms in molecules (AIM) theory were applied to prove the existence of the hydrogen bonds in these clusters. The analysis of the geometries indicated that in the CH...X(X=O, F) type clusters, the proton donor C-H bond in CHF3becomes shorter and the stretching vibrational frequency increases, but in the CH…π type clusters, no obvious rule exist for the changes of the bond length and the vibrational frequency. The natural bond orbital (NBO) analysis was used to help investigate the formation of the hydrogen bond in some systems we studied, and the blue-shifted hydrogen bond may be explained by the dominant rehybridization effect.
coumarin; fluoroform; quantum chemical calculations; binding energy; hydrogen bond
2017-07-03
国家级大学生创新创业训练计划项目(201610449038);滨州学院科研基金项目(2014Y18)
张郅琨(1996—),山东淄博人,在校本科生,化学工程与工艺专业;通讯作者:张丽娟,女,讲师,博士,主要从事理论算研究。
O641.3
A
1008-021X(2017)18-0009-03
(本文文献格式张郅琨,张斌,谭会艳,等.香豆素及其衍生物与CHF3的团簇的理论研究[J].山东化工,2017,46(18):9-11.)
猜你喜欢
大学物理(2022年9期)2022-09-28
疯狂英语·爱英语(2020年6期)2020-07-04
物理通报(2020年7期)2020-07-01
疯狂英语·新策略(2020年6期)2020-06-28
农药科学与管理(2019年8期)2019-11-23
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年6期)2018-07-09
天然产物研究与开发(2018年2期)2018-04-04
原子与分子物理学报(2015年3期)2015-11-24
化工管理(2015年11期)2015-08-15
